
Abstract
Background
Specific HLA-DQ genes have been recognized as necessary - but not sufficient - factors for the occurrence of Celiac Disease (CD). Through a meta-analysis, evaluating the distribution of CD-related HLA genotypes in children, we aimed at providing insights for a potential widened screening strategy.
Methods
After a systematic search on the association between class II HLA genes and CD in children, 46 publications were obtained and assessed for eligibility. A total of 13 eligible studies were submitted to data extraction and analysis (10 case–control studies and 3 cohort studies). Case–control studies collectively enrolled 740 CD patients and 943 controls.
Results
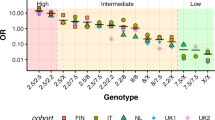
In the population-stratified analysis, the following alleles conferred a significantly increased risk for CD: HLA-DQB1*02 (odds ratio [OR]=10.28) and HLA-DQB1*03:02 (OR=2.24). By drafting a risk gradient to develop CD according to HLA genetic background, the highest risk is confirmed to exist for DQ2/DQ2 homozygous subjects, regardless of the ethnicities (OR=5.4). Actually, the genotype DQ2/β2 showed basically the same risk (OR=5.3). Indeed, no differences have been found in CD risk between DQ2/β2 and DQ2/DQ2, as well as between DQ8/β2 and DQ2/DQ8, and between β2/DQX and DQ2/X.
Conclusion
The HLA-DQB1*02:01 allele is present in more than 90% CD children. In the perspective of a widened pediatric population screening for CD, a double-step process might be suggested: HLA-DQB1*02:01 might be investigated first and, only if this result is positive, children might be candidate for a prospective serologic screening, as a second step.
Similar content being viewed by others

Main
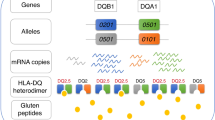
Celiac disease (CD) is a multifactorial immune-mediated disease. The environmental factors triggering most autoimmune disorders are uncertain, as multiple agents—both infectious or not—have been considered. On the contrary, the ingestion of gliadin and some related peptides (sharing a high glutamine and proline content and, thus, called prolamins) has been ascertained to be the critical environmental factor for the development of CD (1, 2). Indeed, the immunopathogenesis of CD recognizes its central event in the specific T-lymphocyte activation by the recognition of complexes, between a class II MHC molecule and a gliadin-derived peptide, which are exposed on the surface of antigen-presenting cells (APCs) in the intestinal mucosa. Such a simple observation clearly suggested the importance of some HLA loci, as the main genetic component of CD (3, 4). Exactly, the genetic predisposition of CD involves class II HLA genes encoding heterodimer MHC II molecules that, in fact, are devoted to the presentation of antigenic peptides (derived from the processing of extracellular antigens by endocytosis, including gliadin) to CD4+ T cells. Carrying two specific MHC class II molecules, called DQ2 and DQ8, is a necessary condition to develop CD during lifetime, although several non-HLA genes are emerging as having a pivotal role in the disease onset and severity, along with some environmental factors (e.g. viral infections, gut microbiota, and others) (5, 6, 7). Each DQ heterodimer is composed of two chains, namely α and β, and the corresponding amino-terminal external domains (α1 and β1) create the peptide-binding cleft, once assembled together (6, 8). DQ2 and DQ8 heterodimers consist of α and β chains encoded by two HLA-DQA1 and two HLA-DQB1 alleles that each individual possesses. Specifically, DQ2 heterodimer includes the α chain encoded by HLA-DQA1*05 allele and the β chain encoded by HLA-DQB1*02 allele. DQ8 heterodimer is encoded by HLA-DQA1*03 and HLA-DQB1*03:02 alleles. In general, it has been demonstrated that more than 90% of CD patients possess HLA-DQ2 and/or HLA-DQ8 alleles, and only less than 2% of CD patients lack these HLA alleles completely (9, 10, 11). HLA-DQ typing has acquired clinical relevance in pediatric gastroenterology: indeed, according to the recent European Society for Pediatric Gastroenterology, Hepatology, and Nutrition (ESPGHAN) guidelines for the diagnosis of CD, the recognition of a predisposing HLA genetics, in association to well-defined clinical and serological criteria, can support the diagnosis without performing any endoscopic procedure to get duodenal biopsies (11, 12). However, this genetic analysis could gain growing attention in the perspective of potential CD-screening programs and strategies in the pediatric age. Most studies investigating the genetic features of CD in humans have been carried out in adults; here, we first performed a meta-analysis evaluating the accuracy of the determination of predisposing HLA genotypes in CD children, in order to find useful insights for the most suitable and cost-effective screening strategy for pediatric CD based upon genetic HLA testing.
Materials and methods
This work is written according to PRISMA guidelines (13).
Protocol
We drew up a protocol including review questions, selection and eligibility criteria, primary and secondary end points, search strategy, methods for data extraction, methods for assessing study quality and risk of bias, strategy for data synthesis, and statistical methodology. On 30 January 2016, the protocol entitled “Association between HLA class II polymorphisms and coeliac disease in infants and children: a meta-analysis” was published in the PROSPERO International prospective register of systematic reviews (PROSPERO 2016:CRD42016033555), and from that date onward, it is available in http://www.crd.york.ac.uk/PROSPERO/display_record.asp?ID=CRD42016033555 (14).
Search Strategy
We performed a systematic search of MEDLINE/PubMed, EMBASE, Web of Science, Cochrane databases, and CINAHL retrieving all publications (case–control study, cross-sectional, and retrospective cohort study) on the association between HLA class II polymorphism and CD in infants and children (<18 years). We searched all English articles published in up to 30 May 2016. An expert librarian performed the search using the following MeSh terms: (‘‘Coeliac Disease) AND (‘‘HLA’’ OR ‘‘human leukocyte antigen’’) AND (‘‘polymorphism’’ OR ‘‘variant’’ OR ‘‘genotype’’ OR “allele”).
Selection criteria were HLA class II genes; any DQ and DR molecules; and celiac disease diagnosed following the clinical criteria: Meeuwisse (1969–1970); Walker-Smith et al. (1990–2012); and Husby et al. (2012 ESPGHAN guidelines) (11, 15, 16).
Risk of Bias (Quality) Assessment
Following a quality-assessment tool for genetic data (Quality Assessment of Genetic Studies in Systematic Reviews, QUAGENS) (17) proposed by our multidisciplinary panel (statisticians, clinical epidemiologists, geneticists, clinicians, and meta-analysts), three pairs of reviewers (one for the clinical criteria, one for laboratory issues, and one for methodology tools) working independently and with adequate reliability verified the following aspects:
-
1
Clinical data: the presence of spectrum disease biases, the possible enrollment of incident or prevalent cases, the inclusion of controls not selected from the same source population, and the occurrence of differential participation in cases and controls.
-
2
Laboratory issues: the misclassification of genotypes or serotypes (including the types and quality of samples, timing of collection, and the method used for typing), the actual laboratory staff blinded to outcome, and the mention of quality controls.
-
3
Methodological features: the possible population stratification, the presence of multiple testing and prestudy odds of true finding (it would be useful interpreting the results in the context of how many polymorphisms have been studied), and the assessment of Hardy-Weinberg equilibrium in controls.
Each question was answered as “yes”, “no”, or “unclear”.
Data Extraction
After a critical reading of the articles, two investigators independently performed data extraction according to the inclusion criteria listed above. The third participant was consulted for discussion to reach an agreement concerning discrepancies. The following items were extracted from each study: first author’s last name, publication date, country of origin, numbers of cases and controls, and typing method.
Data Synthesis and Meta-analysis
STATA 14.2 (StataCorp, College Station, TX, USA) and METADISC 1.4 were used for statistical analysis to perform meta-analysis (18). Heterogeneity was checked by the χ2-test and the I-squared statistics (19). The criteria for identification of heterogeneity were P value less than 0.10 for the χ2-test and an I-squared statistics greater than 50%. When there was no statistical evidence for heterogeneity in effect sizes, we used the fixed-effect model to meta-analyze odds ratios (ORs) or relative risks in probands; when significant heterogeneity was identified, we used the random-effects model and explored sources of significant heterogeneity (20, 21). To take into account the HLA-DQ allele combinations, an individual patient meta-analysis (IPD) was performed considering the copies of DQA1*05:01 and DQB1*02 or DQA1*03:01 and DQB1*03:02 carried by each subject. We termed individuals according to the presence of the HLA-DQA1 and HLA-DQB1 alleles: subjects carrying both the HLA-DQA1*05 and HLA-DQB1*02 alleles are DQ2, subjects carrying both the HLA-DQA1*03 and HLA-DQB1*03:01 alleles are DQ8, those carrying only a single HLA-DQB1*02 allele in the absence of the DQA1*05:01 allele are β2, and those carrying only a single HLA-DQA1*05:01 allele in the absence of the DQB1*02:01 allele are α5. Any other DQ genotypic combination is indicated as DQX. IPD combined all patient data from all studies in one single random multilevel logistic model, taking into account the clustering of patients within studies (20). Furthermore, for each selected study, we calculated sensitivity, specificity, positive likelihood ratio (LR+), and negative likelihood ratio (LR–). In order to produce clinically useful pooled statistics, we meta-analyzed LR+ and LR–. For all estimated, we added their 95% confidence intervals.
Results
Study Selection and Quality of Studies
Our search strategy yielded 1,885 papers for consideration (Figure 1). Following elimination of the 551 duplicates, 1,334 titles and/or abstract were reviewed. Of these, 1,288 were excluded: of the remaining 46 publications, full-text manuscripts were obtained and assessed for eligibility, and other 33 studies were excluded. Finally, a total of 13 studies were deemed eligible for inclusion and were submitted to data extraction and analysis. Studies were published between 1994 and 2016; 10 studies were case–control and three cohort studies (22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34). Case–control studies collectively enrolled 740 CD patients and 943 controls. Study populations were similar with respect to age (median age: 9 [IQR 6–18] in CD and 10 [IQR 8–18] years in controls) and gender (median percentage of males: 46 [IQR 43–48] in CD and 40 [IQR: 33–48] in controls). The quality of studies in terms of laboratory methods description, statistical methodology, and clinical features was mainly low. The majority of studies did not explain at all or were not clear for the majority of items, as shown in Supplementary Figure S1 online.

Flow diagram of the study following the PRISMA statement.
DQ2/DQ8 Heterodimers, HLA-DQB1 and HLA-DQA1 Alleles, and CD Susceptibility (Case–Control Studies)
Individual patient meta-analysis (IPD) showed that each genotype carrying DQ2 or DQ8 heterodimers has an increased risk of developing CD, as it is well known. The maximum risk is represented by DQ2 homozygous state (Supplementary Table S1 online). Furthermore, we analyzed the available data also in the perspective of HLA-DQ alleles rather than in terms of complete heterodimer molecules, namely DQ2 and DQ8. Thus, a gradient for CD risk according to specific HLA-DQB1 and HLA-DQA1 alleles emerged (Supplementary Table S2).
The results displayed in Supplementary Tables S1 and S2 have been summarized in Figure 2, representing the spectrum of risk to develop CD in children, based on IPD meta-analysis data. The highest risk is confirmed to exist for DQ2/DQ2 homozygous subjects (namely, carrying HLA-DQA1*05 and HLA-DQB1*02 on both respective loci), also by considering all the ethnicities. These children have an odds ratio (OR) of 5.4 (95% confidence interval, 95%CI: 4.1–6.8) to develop CD, compared with the reference risk associated with any HLA-DQ genotype other than those coding any chain (α or β) of DQ2 or DQ8 heterodimers. Actually, the genotype DQ2/β2 consisting of two DQB1*02 alleles and only one HLA-DQA1*05 allele (with the other HLA-DQA1 of any different type) has basically the same risk (OR=5.3; 95%CI:4.1–6.5). Indeed, no differences have been found in CD risk between DQ2/β2 and DQ2/DQ2 (χ2=0.06, P=0.8089), as well as between DQ8/β2 and DQ2/DQ8 (χ2=0.19, P=0.6616), and between β2/DQX and DQ2/X (χ2=0.26, P=0.6071).

Risk gradient for pediatric CD, according to HLA-DQ genotype. CD, celiac disease; HLA, human leukocyte antigen.
Then, starting from this peak of risk to develop pediatric CD, a spectrum of decreasing risk was drafted, as it follows:
-
DQ8/β2 children being HLA-DQA1*03 and DQB1*03:02 heterozygous, but having also the allele coding for the β chain of DQ2 heterodimer (OR=4.6; 95%CI: 2.8–6.3);
-
DQ2/DQ8 and DQ2/DQX children, namely DQ2 (DQA1*05 and DQB1*02) heterozygous, regardless of the presence of the DQ8 genotype (OR=4.1, 95% CI: 2.5–5.8 and OR 4.1, 95% CI 3.0–5.1, respectively);
-
β2/DQX children being only heterozygous for DQ2 β chain (DQB1*02), without any other alleles coding for chains of DQ2 or DQ8 heterodimers (OR=3.9; 95% CI 2.7–5.0);
-
DQ8/DQX children being basically only DQ8 heterozygous (OR=2.8; 95% CI: 1.6–3.9);
-
α5/DQX children carrying only HLA-DQA1*05 allele, without any allele coding for other chains of DQ2 or DQ8 heterodimers (OR=2.2; 95% CI 0.1–4.4).
No enough data were available to calculate OR for the remaining combinations of HLA-DQ alleles, such as genotypes including only DQB1*03:02 (namely, encoding only the β chain of DQ8 heterodimer) or those consisting of both DQB1*02 alleles without any DQA1 allele to complete any of DQ2 or DQ8 heterodimers. Finally, we reported in Supplementary Table S3 the genotypes of children diagnosed with CD from three studies of this meta-analysis that provided complete data about HLA-DQ alleles. By paying attention to the presence of HLA-DQB1 alleles, 96.8% of pediatric CD genotypes can be defined by the presence of at least one allele coding the β chain of DQ2 or DQ8 heterodimer. In other words, only 3.2% of these CD children found their genetic predisposition in other alleles than those coding DQ2 or DQ8 β chains: 0.5% CD children were carrying the HLA-DQA1*05 allele only and the remaining 2.7% showed allelic variants being different from those of DQ2 and DQ8 heterodimers.
CD Susceptibility and HLA-DQB1 Alleles (Case–control Studies)
Following the previous results and observations on the pediatric CD risk according to the genotype, we focused our analysis on the data regarding HLA-DQB1 alleles. Among the 10 case–control articles selected for the meta-analysis, seven articles reported the frequency of the HLA-DQB1*02 allele (one in Asian and Amerindian, two in Arabic, and three in Caucasian populations), whereas five articles reported the frequency of the HLA-DQB1*03:02 allele (one in Asian, Arabic, and Amerindian and two in Caucasian populations). 300 out of 390 CD patients (77%) vs. 226 out of 611 (37%) healthy controls carried HLA-DQB1*02, whereas 86 out of 311 patients (28%) vs. 84 out of 423 (20%) healthy controls carried HLA-DQB1*03:02. In the population-stratified analysis, these alleles significantly conferred an increased risk for CD: HLA-DQB1*02 (OR: 10.28, 95%IC: 4.49–23.5), as shown in Figure 3a, and HLA-DQB1*03:02 (OR: 2.24; 95%IC: 1.02–4.91), as shown in Figure 3b. Because of the prevalent influence of carrying at least HLA-DQB1*02 allele for pediatric CD development, we meta-analyzed the LR+ and LR− of CD in children according to the presence of HLA-DQB1*02 allele. All studies provided a very high LR−, whereas LR+ resulted to be low and variable among different studies. The pooled estimate of LR+ was 2.55 (IC 0.97–6.72), whereas LR− showed a good discriminatory power: 0.31 (IC 0–17–0.56), as shown in Figure 4a. Egger’s regression test showed no evidence of publication bias (Egger’s regression test P values >0.1). Finally, we meta-analyzed the LR+ and LR− of CD if carrying one of two alleles that codifies for β chains of DQ2 or DQ8 molecules, namely HLA-DQB1*02 or HLA-DQB1*03:02 (Figure 4b). The pooled estimate of LR+ was quite low: 1.98 (IC 0.97–3.98), whereas LR− showed a good discriminatory power: 0.13 (IC 0–04–0.37). Egger’s regression test showed no evidence of publication bias (Egger’s regression test P values >0.1).

Population-stratified CD risk analysis according to HLA-DQB1 alleles. OR values for HLA-DQB1*02 (a) and HLA-DQB1*03:02 (b). CD, celiac disease; HLA, human leukocyte antigen.

LR of CD according to HLA genotype. Positive and negative LR according to the presence of HLA-DQB1*02 allele (a) or to the presence of one of two alleles that codifies for β chains of DQ2 or DQ8 molecules, namely HLA-DQB1*02 or HLA-DQB1*03:02 (b). CD, celiac disease; HLA, human leukocyte antigen; LR, likelihood ratio.
CD Susceptibility and HLA-DQB1 Alleles (Retrospective Cohort Studies)
Three cohort studies (all from Caucasian population), involving 2,238 subjects carrying HLA-DQB1*02, 2,110 carrying DQB1*03:02, 593 carrying both, and 6,198 subjects without risk alleles, were retrieved. Relative risk with respect to subjects without risk alleles is summarized in Supplementary Table S4.
Discussion
This is the first pediatric meta-analysis reviewing the disease association of HLA-DQB1 and HLA-DQA1 alleles encoding DQ2 and DQ8 heterodimers predisposing to CD. It included 13 pediatric studies between 1994 and 2016 adhering to all established criteria, providing 740 CD pediatric patients and 943 controls for statistical analysis. Here, the increase in the risk to develop CD in children carrying DQ2 and/or DQ8 molecules has been fully confirmed, of course.
As explained in the Introduction, the predisposing DQ2 and DQ8 heterodimers are composed of the association of α and β chains, encoded by HLA-DQA1*05 and DQB1*02 alleles and HLA-DQA1*03:01 and DQB1*03:02 alleles, respectively (11). However, our meta-analysis showed the strongest genetic association with pediatric CD for those alleles encoding the β chains of susceptibility heterodimers and, particularly, for HLA-DQB1*02:01 allele encoding DQ2 β chain. In other words, HLA genotypes coding for DQ2 or DQ8 heterodimers, but also those including only the alleles of the respective β chains (regardless of the concomitant presence of DQ2 or DQ8 α chains) have an increased risk of developing CD.
Recently, Megiorni et al. reviewed the role of HLA-DQA1 and HLA-DQB1 in the predisposition to CD and put forward a picture of HLA-DQ status and risk of CD, where DQ2/DQ8 heterozygous and DQ2 homozygous individuals showed a very high risk and, to follow, DQ8 homo- and heterozygous along with DQ2 heterozygous and people carrying a double dose of DQB1*02 (neither having the whole DQ2 nor DQ8 heterodimer) resulted to have a high risk, anyway (35). Our meta-analysis supports these conclusions, as summarized in Figure 2. Here, it is shown that a double dose of HLA-DQB1*02 is associated with the highest risk to develop pediatric CD (OR>5) and that also a single dose of HLA-DQB1*02 leads to a high risk (OR being around 4). Importantly, based on our statistical results, children having only one HLA-DQB1*02 allele (without any other HLA-DQ2 or HLA-DQ8 allele) showed basically the same risk to develop CD as children carrying heterozygous HLA-DQ2 and/or HLA-DQ8 full genotypes. Moreover, the results of our meta-analysis are in a great agreement with the original research by Megiorni et al., including 437 Italian children with CD, in addition to 551 controls, who were HLA-DQ-genotyped. They calculated a gradient of CD risk by evaluating unrounded case–control DQ frequencies and considering a disease prevalence of 1:100. In summary, they found a disease risk of 1:26 for children being homozygous for HLA-DQB1*02, despite the absence of the other genes coding for DQ2 or DQ8. This result is very interesting, if it is compared with the double heterozygous DQ2/DQ8, the heterozygous DQ2 being HLA-DQB1*02 homozygous, and the DQ8 heterozygous having one HLA-DQB1*02 allele, which showed a disease risk of 1:7, 1:10, and 1:24, respectively (36).
Indeed, adult and pediatric CD are considered to represent the phenotypic spectrum of the same disease, as concluded in a very recent research by Senapati et al., which represents the first comparative study of genetic markers, including HLA-DQ2 and HLA-DQ8 haplotypes, between children and adults affected with CD. Exactly, these authors confirmed that both children and adults are permitted to develop CD by HLA-DQ2 and HLA-DQ8 haplotypes; moreover, through the analysis of the genotype data set acquired by using Immunochip, their analysis also supported the conclusion that different sets of non-HLA loci determine the disease heterogeneity (including the age of onset, the disease severity, and the occurrence of extraintestinal clinical manifestations) among people sharing that same genetic HLA-DQ background (37).
This finding can be translated in the evidence-based medicine by considering the LR, as we made in this meta-analysis. LR combines the sensitivity and specificity of the test (HLA-DQ genotyping, in our case) to determine whether a test result changes the probability that a condition, like a disease state (pediatric CD, in our case), exists (38).
Therefore, we made the meta-analysis of LR+ and LR– to develop pediatric CD, if carrying one of two alleles that codifies for β chains of DQ2 or DQ8 molecules, namely HLA-DQB1*02 or HLA-DQB1*03:02 (Figure 4b). The pooled estimate of LR+ was quite low, whereas LR– showed a good discriminatory power, confirming once more that HLA testing is accurate to rule out pediatric CD, but does not provide final confirmation for the diagnosis, of course. Interestingly, we obtained such a result by considering only the β chain genotype, namely HLA-DQB1 alleles, regardless of the association to HLA-DQA1 alleles coding for α chains of DQ2 and DQ8 heterodimers, being HLA-DQA1*05 and HLA-DQA1*03:01, respectively.
Our meta-analysis, along with these studies, may represent an epidemiological confirmation of the biological data supporting the preponderant role of class II MHC β chain and, particularly, of that codified by DQB1*02 allele, in the immunological pathogenesis of pediatric CD. Nenna et al. demonstrated that antitransglutaminase (anti-tTG) autoantibody titers were significantly different in their CD patients receiving a gluten-containing diet, according to the HLA-DQB1*02 status; moreover, patients with at least one HLA-DQB1*02 allele resulted to have more expressed clinical and histological CD (39). Under the molecular perspective, the relationship between the magnitude of gluten-specific T-cell responses and the dose effect of HLA-DQ2 genes has been reported since the beginning of the century (40). Moreover, further studies demonstrated that the high content of proline and glutamine residues of HLA-DQ2-restricted gliadin epitopes (lending important biological properties, such as the resistance to gastrointestinal proteolysis and the facility to be deamidated by tTG with enhanced immunogenicity) is fundamental for the interaction with class II MHC molecules (41). On the basis of X-ray crystallography research, it has been suggested that some DQ2 β chain residues, participating in the peptide-binding cleft (particularly Arg-β70 and Lys-β71 of β chain encoded by HLA-DQB1*02), are responsible for the interaction with several residues of gliadin epitope and, thus, may be essential in the pathogenesis of CD (42).
Thus, the risk stratification emerging from our meta-analysis confirmed the great importance of allelic HLA-DQ status and, particularly, of the “dose” of DQB1*02 allele (rather than the presence of whole heterodimer molecules DQ2/DQ8) in the predisposition to develop CD in the pediatric population and, importantly, regardless of the ethnicity. Indeed, the selected studies regarded different ethnicities, including Asian, Arabic, and Amerindian children, in addition to Caucasian subjects. According to the growing diagnostic recognition of CD in pediatric populations from non-western countries, seven of our selected studies were from outside Europe and North America (South America (1), Middle East (3), North Africa (2), and India (1)). Thus, an added value of this meta-analysis (based upon those eligible studies including children only) is that such a HLA-DQ genetic background of pediatric CD predisposition was similar regardless of the ethnicity. So far, most clinical studies concerning CD mainly included Caucasian adult patients, and were settled in western countries. In Europe, the overall prevalence is estimated to be around 1%, ranging from 0.3% (Germany) to 2.4% (Finland). However, CD seems to be as much prevalent outside Europe, as similar prevalence was described both in North and South America and even in non-Caucasian populations settled in India, in the Middle East, and in North Africa (43, 44, 45).
Finally, as mentioned in the Introduction, the most recent ESPGHAN guidelines for the diagnosis of pediatric CD significantly increased the practical usefulness of HLA typing: indeed, those introduced the possibility to achieve the final diagnosis of CD in pediatric patients without performing intestinal biopsies in selected cases showing specific clinical and serological criteria and, importantly, carrying HLA alleles encoding DQ2 and/or DQ8 heterodimers (11). Moreover, the growing interest in CD case finding strategies in at-risk groups and also in the general pediatric population might further increase the request of these genetic analyses, which are currently limited by the traditional costs of high-resolution HLA typing (46, 47). According to current clinical studies, most children with CD carry DQ2 and, in a lesser extent, DQ8 heterodimers; the remaining part (being DQ2 and DQ8 negative) mainly consists of children carrying one or two HLA-DQB1*02 alleles or, in a much lesser extent, only the HLA-DQA1*05 allele (coding the α chains of HLA-DQ2). Less than 1% of children affected with CD resulted to lack the aforementioned HLA alleles completely (35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48). Generally, HLA typing is quite expensive and, thus, new techniques to reduce its costs are being in the experimental phase (49, 50).
However, the knowledge that HLA-DQB1*02 allele is a major HLA risk factor being present in almost all CD children might be considered in the perspective of a widened pediatric population screening assessing the genetic risk of CD in children, as a first step. Very recently, Bjorck et al. published their cohort study including 13,860 HLA-genotyped subjects at birth and prospectively screened for the occurrence of CD: ~80% of 9-year-old children resulted to carry HLA-DQB1*02 and the remaining part showed HLA-DQB1*03:02 (47). In a less recent study by Perez-Bravo F et al., including 62 Chilean CD children, it is evident that only 6% of their patients lacked HLA-DQB1*02 or HLA-DQB1*03:02 alleles (24). Similar observations emerged from the Italian study by Congia M et al., including 62 CD patients (25). Importantly, this meta-analysis provided tailored data about HLA-DQ genotypes for the pediatric population affected with CD all over the world. The greatest importance of DQ2 heterodimer and, in particular, of HLA-DQB1*02:01 allele (recovered in more than 90% of CD children, at least in a single copy) in the determination of the genetic susceptibility to CD for children, regardless of their ethnicity, has been fully confirmed by our meta-analysis.
Thus, we consider that children might be screened in a double-step process: they might be first investigated for the presence of HLA-DQB1*02 allele, which is enough to label a patient as being predisposed to CD and, importantly, is possessed by the vast majority of CD children, regardless of the ethnicity. Thus, children who are identified as being predisposed to CD after this first step might be taken in account for a follow-up during the time by the use of serological investigations, according to the current clinical recommendations.
References
Ludvigsson JF, Bai JC, Biagi F et al. Diagnosis and management of adult coeliac disease: guidelines from the British Society of Gastroenterology. Gut 2014;63:1210–28.
Vader W, Kooy Y, Van Veelen P et al. The gluten response in children with celiac disease is directed toward multiple gliadin and glutenin peptides. Gastroenterology 2002;122:1729–37.
Arentz-Hansen H, McAdam SN, Molberg Ø et al. Celiac lesion T cells recognize epitopes that cluster in regions of gliadins rich in proline residues. Gastroenterology 2002;123:803–9.
Sollid LM, Jabri B . Triggers and drivers of autoimmunity: lessons from coeliac disease. Nat Rev Immunol 2013;13:294–302.
Lionetti E, Castellaneta S, Francavilla R et al. Introduction of gluten, HLA status, and the risk of celiac disease in children. N Engl J Med 2014;371:1295–303.
Hardy MY, Tye-Din JA . Coeliac disease: a unique model for investigating broken tolerance in autoimmunity. Clin Transl Immunol 2016;5:e112.
Lebwohl B, Ludvigsson JF, Green PH . The unfolding story of celiac disease risk factors. Clin Gastroenterol Hepatol 2014;12:632–5.
Abbas AK, Lichtman AH, Pillai S. The major histocompatibility complex. eds. Cellular and molecular Immunology, 6th edn, 2014: 97–110.
Di Sabatino A, Corazza GR . Coeliac disease. Lancet 2009;373:1480–93.
Tjon JM, van Bergen J, Koning F . Celiac disease: how complicated can it get? Immunogenetics 2010;62:641–51.
Husby S, Koletzko S, Korponay-Szabó IR et al. European Society for Pediatric Gastroenterology, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease. J Pediatr Gastroenterol Nutr 2012;54:136–60.
Garnier-Lengliné H, Cerf-Bensussan N, Ruemmele FM . Celiac disease in children. Clin Res Hepatol Gastroenterol 2015;39:544–51.
Moher D, Liberati A, Tetzlaff J, Altman DG, PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. J Clin Epidemiol 2009;62:1006–12.
PROSPERO International prospective register of systematic reviews published the protocol “Association between HLA class II polymorphisms and coeliac disease in infants and children: a meta-analysis”; (http://www.crd.york.ac.uk/PROSPERO/display_record.asp?ID=CRD42016033555) Accessed 30 January 2016.
Meeuwisse GW . Round table discussion. Diagnostic criteria in coeliac disease. Acta Paediatr Scand 1970;59:461–3.
Walker-Smith JA, Guandalini S, Schmitz J . Revised criteria for diagnosis of coeliac disease. Arch Dis Child 1990;65:909–11.
De Silvestri A, Capittini C, Pasi A. QUAGENS: a tool for QUality Assessment of GENetic Studies in systematic reviews. A proposal, 2016 (http://evidencelive.org/wp-content/uploads/2016/06/DE-SILVESTRI-22.pdf).
Zamora J, Abraira V, Muriel A et al. Meta-DiSc: a software for meta-analysis of test accuracy data. BMC Med Res Methodol 2006;6:31.
Higgins JP, Thompson SG . Quantifying heterogeneity in a meta-analysis. Stat Med 2002;21:1539–58.
Der Simonian R, Laird N . Meta-analysis in clinical trials. Control Clin Trials 1986;7:177–88.
Higgins JPT, Green S. edsCochrane Handbook for Systematic Reviews of Interventions, Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. (http://www.cochrane-handbook.org) Accessed 1 March 2015.
Alarida K, Harown J, Di Pierro MR et al. HLA-DQ2 and -DQ8 genotypes in celiac and healthy Libyan children. Dig Liver Dis 2010;42:425–7.
Björck S, Brundin C, Lörinc E et al. Screening detects a high proportion of celiac disease in young HLA-genotyped children. J Pediatr Gastroenterol Nutr 2010;50:49–53.
Pérez-Bravo F, Araya M, Mondragón A et al. Genetic differences in HLA-DQA1* and DQB1* allelic distributions between celiac and control children in Santiago, Chile. Hum Immunol 1999;60:262–7.
Congia M, Cucca F, Frau F et al. A gene dosage effect of the DQA1*0501/DQB1*0201 allelic combination influences the clinical heterogeneity of celiac disease. Hum Immunol 1994;40:138–42.
El-Akawi ZJ, Al-Hattab DM, Migdady MA . Frequency of HLA-DQA1*0501 and DQB1*0201 alleles in patients with coeliac disease, their first-degree relatives and controls in Jordan. Ann Trop Paediatr 2010;30:305–9.
Kaur G, Sarkar N, Bhatnagar S et al. Pediatric celiac disease in India is associated with multiple DR3-DQ2 haplotypes. Hum Immunol 2002;63:677–82.
Krini M, Chouliaras G, Kanariou M et al. HLA class II high-resolution genotyping in Greek children with celiac disease and impact on disease susceptibility. Pediatr Res 2012;72:625–30.
Laadhar L, Toumi A, Kallel-Sellami M et al. HLA class II polymorphism in children with coeliac disease in Tunisia: is there any influence on clinical manifestation? Eur J Gastroenterol Hepatol 2009;21:1286–90.
Mäki M, Mustalahti K, Kokkonen J et al. Prevalence of Celiac disease among children in Finland. N Engl J Med 2003;348:2517–24.
Tüysüz B, Dursun A, Kutlu T et al. HLA-DQ alleles in patients with celiac disease in Turkey. Tissue Antigens 2001;57:540–2.
Mustalahti K, Sulkanen S, Holopainen P et al. Coeliac disease among healthy members of multiple case coeliac disease family. Scand J Gastroenterol 2002;2:161–5.
Hernández-Charro B, Donat E, Miner I et al. Modifying effect of HLA haplotypes located trans to DQB1*02-DRB1*03 in celiac patients of Southern Europe. Tissue Antigens 2008;71:213–8.
Çakır M, ; Baran M, Uçar F et al. Accuracy of HLA-DQ genotyping in combination with IgA anti-tissue transglutaminase serology and a "scoring system" for the diagnosis of celiac disease in Turkish children. Turk J Pediatr 2014;56:347–453.
Megiorni F, Pizzuti A . HLA-DQA1 and HLA-DQB1 in Celiac disease predisposition: practical implications of the HLA molecular typing. J Biomed Sci 2012;19:88.
Megiorni F, Mora B, Bonamico M et al. HLA-DQ and risk gradient for celiac disease. Hum Immunol 2009;70:55–9.
Senapati S, Sood A, Midha V et al. Shared and unique common genetic determinants between pediatric and adult celiac disease. BMC Med Genomics 2016;9:44.
Pauker SG, Kassirer JP . Therapeutic decision-making: a cost-benefit analysis. N Engl J Med 1975;293:229–34.
Nenna R, Mora B, Megiorni F et al. HLA-DQB1*02 dose effect on RIA anti-tissue transglutaminase autoantibody levels and clinicopathological expressivity of celiac disease. J Pediatr Gastroenterol Nutr 2008;47:288–92.
Vader W, Stepniak D, Kooy Y et al. The HLA-DQ2 gene dose effect in celiac disease is directly related to the magnitude and breadth of gluten-specific T cell responses. Proc Natl Acad Sci USA 2003;100:12390–5.
Sollid LM . Coeliac disease: dissecting a complex inflammatory disorder. Nat Rev Immunol 2002;2:647–55.
Kim CY, Takahashi K, Nguyen TB et al. Identification of a nucleic acid binding domain in eukaryotic initiation factor eIFiso4G from wheat. J Biol Chem 1999;274:10603–8.
Mustalahti K, Catassi C, Reunanen A et al. The prevalence of celiac disease in Europe: results of a centralized, international mass-screening project. Ann Med 2010;42:587–95.
Catassi C, Anderson RP, Hill ID et al. World perspective on celiac disease. J Pediatr Gastroenterol Nutr 2012;55:494–9.
Yuan J, Gao J, Li X et al. The tip of the "celiac iceberg" in China: a systematic review and meta-analysis. PLoS ONE 2013;8:e81151.
Mitchell RT, Sun A, Mayo A et al. Coeliac screening in a Scottish cohort of children with type 1 diabetes mellitus: is DQ typing the way forward? Arch Dis Child 2016;101:230–3.
Björck S, Lynch K, Brundin C et al. Repeated screening can be restricted to at-genetic-risk birth cohorts. J Pediatr Gastroenterol Nutr 2016;62:271–5.
Stanković B, Radlović N, Leković Z et al. HLA genotyping in pediatric celiac disease patients. Bosn J Basic Med Sci 2014;14:171–6.
Monsuur AJ, de Bakker PI, Zhernakova A et al. Effective detection of human leukocyte antigen risk alleles in celiac disease using tag single nucleotide polymorphisms. PLoS ONE 2008;3:e2270.
Elias J, Hoorweg-Nijman JJ, Balemans WA . Clinical relevance and cost-effectiveness of HLA genotyping in children with Type 1 diabetes mellitus in screening for coeliac disease in the Netherlands. Diabet Med 2015;32:834–838.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
The first two authors contributed equally to this work.
Statement of Financial Support
This study was supported by the Fondazione IRCCS Policlinico San Matteo (Pavia, Italy), through research funds of the project no. 870-rcr2016-50.
Supplementary material is linked to the online version of the paper at
Rights and permissions
About this article

Cite this article
De Silvestri, A., Capittini, C., Poddighe, D. et al. HLA-DQ genetics in children with celiac disease: a meta-analysis suggesting a two-step genetic screening procedure starting with HLA-DQ β chains. Pediatr Res 83, 564–572 (2018). https://doi.org/10.1038/pr.2017.307
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2017.307
This article is cited by
-
Screening for celiac disease in juvenile idiopathic arthritis
Pediatric Rheumatology (2022)
-
Associations between human leukocyte antigens and renal function
Scientific Reports (2021)
-
HLA class II genes in precision-based care of childhood diseases: what we can learn from celiac disease
Pediatric Research (2021)
-
Frequency of HLA celiac disease risk alleles and haplotypes in healthy adults in Tamil Nadu
Indian Journal of Gastroenterology (2019)
-
Individual screening strategy for pediatric celiac disease
European Journal of Pediatrics (2018)

{kind=link}