
Abstract
Background
The objective of our study was to examine the risk for submicroscopic chromosomal aberrations among fetuses with apparently isolated solitary kidney.
Methods
Data acquisition was performed retrospectively by searching Israeli Ministry of Health-computerized database. All cases having chromosomal microarray analysis (CMA), referred because of an indication of isolated unilateral kidney agenesis between January 2013 and September 2016, were included. Rate of clinically significant CMA findings in these pregnancies was compared to pregnancies with normal ultrasound, based on a systematic review encompassing 9,792 cases and local data of 5,541 pregnancies undergoing CMA because of maternal request.
Results
Of the 81 pregnancies with isolated solitary kidney, 2 (2.47%) loss-of-copy number variants compatible with well-described deletion syndromes were reported (16p11.2–16p12.2 and 22q11.21 microdeletion syndromes). In addition, one variant of unknown significance was demonstrated. The relative risk for pathogenic CMA findings among pregnancies with isolated unilateral renal agenesis was not significantly different compared with the control population.
Conclusion
CMA analysis in pregnancies with unilateral renal agenesis might still be useful, to the same degree as it can be in the general population.
Similar content being viewed by others

Main
Congenital anomalies of the kidney and urinary tract (CAKUT) occur in three to six of 1,000 live births, representing ~20% of the prenatally detected sonographic anomalies (1, 2). Unilateral renal agenesis is a relatively frequent anomaly of the CAKUT spectrum, diagnosed in ~500–2,000 live births (3). This malformation is usually asymptomatic postnatally and detected incidentally.
Prenatal nonvisualization of fetal kidney is related to renal ectopia (pelvic kidney) in almost half of the cases (4). True absent kidney is usually related to renal agenesis, whereas some cases result from an involution of multicystic dysplastic kidney (5). Evaluation of pregnancies with empty renal fossa includes a detailed anatomic survey for associated anatomic anomalies. Additional urological abnormalities can be demonstrated in about half of the cases, including vesicoureteral reflux and urinary tract obstructions (6). Associated nonurological anomalies are found in around one-third of the cases, such as cardiac malformations, genital tract anomalies, skeletal defects, and gastrointestinal tract malformations (3).
Genetic factors have a major role in the pathogenesis of CAKUT (7). About 10% are related to single-gene mutations (8), whereas about 16% are associated with copy number variants (CNVs), such as chromosome 17q12 and 22.q11.2 microdeletions. Several reports of familial renal agenesis have been described, suggestive of various inheritance types, most cases consistent with autosomal-dominant inheritance with incomplete penetrance, and variable expressivity (9). Renal agenesis, especially bilateral, has been associated with several chromosomal aberrations. Nicolaides et al. reported a 5% rate of aneuploidy in prenatally detected isolated bilateral renal agenesis; in one of 19 fetuses, a 47,XYY karyotype was demonstrated (10). However, in the three pregnancies with isolated unilateral renal agenesis, karyotype testing yielded normal results. Westland et al. performed high-density single-nucleotide polymorphism microarray in 80 patients with solitary functioning kidney, and identified known or novel potentially pathogenic genomic imbalances in 14% of the cases (11). However, 26 (30%) of these patients presented additional structural or neurocognitive disorders.
Because of increased risk of associated anomalies, the evaluation of pregnancies with kidney agenesis may include invasive prenatal testing with chromosomal microarray analysis (CMA). Nonetheless, and to the best of our knowledge, no reports have been published yet for examining the rates of abnormal CMA results in pregnancies with solitary kidney. Thus, the objective of our study was to shed light on this issue.
Methods
A retrospective cohort study examined that all CMA analyses were reported to Israeli Ministry of Health-computerized database between January 2013 and September 2016. Routine pregnancy follow-up includes nuchal translucency measurement at 11–14 weeks, as well as subsequent detailed fetal anatomic survey and biochemical tests for Down’s syndrome screening. In Israel, pregnancies with sonographic anomalies, excluding fetal soft markers for chromosome anomalies, are referred for genetic counseling and advised to undergo invasive prenatal testing for CMA analysis, which is financed by the Ministry of Health. The database included indication for CMA testing, maternal age, timing of sonographic anomaly diagnosis, and CMA result. DNA extraction was performed in standard techniques. CMA analyses were performed using either single-nucleotide polymorphism-based array or comparative genomic hybridization. All tests were provided by clinical laboratories and approved by the Israeli Ministry of Health. To promote consistency in interpretation and reporting of genomic microarray results, all standards and reports were based on the recommendation of the Israeli Society of Medical Genetics. These recommendations in turn rely on the American College of Medical Genetic and Genomics guidelines (12, 13). In short, a pathogenic CNV was reported when the CNV was documented as clinically significant in multiple peer-reviewed publications. This category includes large CNVs, which may not be described in the medical literature at the size observed in the patient, but overlap a smaller interval with clearly established clinical significance. In addition, cytogenetically visible alterations (3–5 Mb) were defined as pathogenic, unless well-established polymorphism of the particular region was described (13). Variants were defined as variants of unknown significance when, at the time of reporting, insufficient evidence was available for unequivocal determination of clinical significance (e.g., when no clear evidence was found for the pathogenicity of a dosage effect for gene/s included in the CNV), and the variant met the reporting criteria established by the laboratory (13). Of note, according to the Israeli Medical Genetics Association guidelines, variants of unknown significance, which include loss of copy number larger than 1 Mb or gain of copy number larger than 2 Mb should be reported.
All pregnancies with diagnosis of isolated renal agenesis and documentation of CMA result were included in the analysis. We excluded pregnancies with additional abnormal sonographic anomalies (including soft signs). Genetic counseling was performed in all cases. The study was approved by the Institutional Review Board Committee (Helsinki Committee) for Human Subjects.
Copy number gains or losses were analyzed by several public databases (UCSC Genome Browser: http://www.genome.ucsc.edu/cgi-bin/hgGateway, DECIPHER database: http://decipher.sanger.ac.uk/, and ClinGen database: https://www.clinicalgenome.org/). CNVs were categorized as benign, variants of unknown significance, or pathogenic according to the American College of Medical Genetics standards and guidelines (12, 13).
Rate of clinically significant CMA findings in pregnancies with isolated renal agenesis was compared with a control population of 9,272 pregnancies, based on a systematic review by Callaway et al. (14). These cases comprise 5,108 pregnancies referred because of advanced maternal age, and 4,164 cases in “other ascertainment group” (mostly parental request and abnormal serum-screening result). In this population, 94 (1.03%) clinically significant CNV were found. In addition, we used a group of 5,541 pregnancies with normal ultrasound, undergoing CMA due to maternal request in one large Israeli medical center. In this group, CMA testing yielded a 1.39% rate of abnormal results (77 cases, unpublished data), in the range of previously reported 0.5–1.7% estimate.
Statistical analysis was performed using SPSS software (SPSS, Chicago, IL). Continuous variables were presented as averages with SDs. Categorical data were presented as numbers and proportions (absolute risks) with 95% confidence intervals (CI). The effect estimates were calculated as relative risk (RR) with 95% CI. P<0.05 was considered statistically significant for all comparisons.
Results
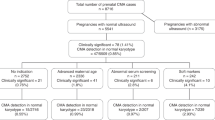
Over the years 2013–2016, 6,046 CMA analyses were reported to the Ministry of Health. Of these, 81 cases were referred because of isolated renal agenesis. Mean maternal age (when reported) was 33.9±3.1 years, not significantly different from the entire cohort (31.7±5.6 years old). The timing of the sonographic diagnosis was 20.8±5.1 weeks (when reported, 34 cases), earlier than that of the entire cohort (22.9±6.9 weeks, P=0.034).
CMA analysis results are summarized in Table 1. In two cases (absolute risk 2.47%, 95% CI 0.2–9.1%), CMA detected loss of CNVs, consistent with well-described deletion syndromes. In addition, one variant of unknown significance was observed (absolute risk 1.2%, 95% CI 0.01–7.3%), inherited from a healthy mother.
The RR for pathogenic CMA findings among pregnancies with isolated unilateral renal agenesis was calculated to be 1.78 (95% CI 0.44–7.10). This was compared with the risk for abnormal CMA findings in a control population of 9,272 pregnancies. This was compared to the risk for abnormal CMA findings in a control population of 9272 pregnancies (14), yielding a non-significant risk difference for abnormal CMA findings (RR 2.44 (95% CI 0.61–9.79), p=0.197). Comparison of local control population of 5,541 pregnancies with normal ultrasound also did not demonstrate statistical significance (RR 1.78 (95% CI 0.44–7.10, P=0.41)).
Discussion
In our cohort, we detected two losses of CNVs with isolated unilateral renal agenesis that are consistent with well-described deletion syndromes. In both cases, an apparently isolated renal defect was actually related to a more complex clinical condition.
The proximal short arm of chromosome 16 involving bands 16p11.2–16p12.2 is a region enriched with repetitive sequence elements that predispose to several reciprocal deletions and duplications (15). These include 220-kb microdeletions encompassing the SH2B1 gene, associated with an isolated early-onset severe obesity as well as obesity with developmental delay (OMIM #613444). The common microdeletions/duplications at 16p11.2 sized ~600 kb are associated with microcephaly, neurocognitive difficulties, and obesity. Finally, the locus includes microscopically visible duplications at 16p11.2–16p12.1/2 sized 7–9 Mb, related to an increased risk for developmental delay and autism, and reciprocal microdeletions similar to those shown in our study.
A phenotype–genotype correlation for de novo-visible interstitial chromosome deletion 16p11.2 was first described by Hernando et al. in 2002 (ref. 16). The authors reported a male infant with multiple congenital anomalies, including dysmorphic features, tetralogy of Fallot, pulmonary atresia, severe growth retardation, cryptorchidism, and unilateral renal agenesis. In 2014, Okamoto et al. reported a patient with a 16p12.2–p11.2 deletion and a patient with a 16p12.2–p11.2 duplication, and reviewed the reports of the two syndromes, by comparing the clinical manifestations (17). The authors summarized the clinical findings in eight patients with 16p12.2–p11.2 deletions sized 7.1–8.7 Mb. Common features included intellectual disability, flat face, low-set malformed ears, significant delay in motor and speech development, feeding difficulties, and recurrent ear infections. No additional renal anomalies were noted.
In our report, we describe a fetus with isolated renal agenesis and 16p12.2–p11.2 deletion. Therefore, it might be that unilateral renal agenesis is a rare finding of 16p12.2–p11.2 deletion syndrome.
The second abnormal CMA result in our analysis is a maternally inherited 22q11.21 deletion sized 3.16 Mb, consistent with DiGeorge/velocardiofacial syndrome (OMIM#188400). This condition is one of the most common chromosomal microdeletion disorders, occurring in about one in every 4,000–6,000 live births (18). According to some reports, the syndrome can be diagnosed in one in every 1,000 seemingly anatomically normal fetuses (19). The syndrome is characterized by a wide range of abnormal findings, including congenital heart defects (especially conotruncal defects), palatal anomalies (particularly velopharyngeal insufficiency), hypocalcemia, immune deficiency, learning difficulties, and typical facial features (20). Various urinary tract anomalies have been reported in about one-third of these patients (18, 21, 22). Specifically, kidney agenesis was noted in 7 of 80 patients with chromosome 22q11.2 microdeletion (23). However, a collaborative survey of 272 fetuses with 22q11.2 deletion has demonstrated that all pregnancies with kidney abnormalities were combined with other sonographic abnormalities (22). Nevertheless, the association between 22q11.21 deletion and unilateral renal agenesis in our study might not be incidental.
In case 3, gain of CNV in chromosomal region 2q11.1–11.2 sized 1.49 Mb was found. This region encompasses five OMIM-morbid genes, including ADRA2B (OMIM #104260), associated with autosomal dominant familial adult myoclonic epilepsy type 2; TMEM127 (OMIM #613403), related to autosomal dominant susceptibility to pheochromocytoma; SNRNP200 (OMIM #610359), associated with autosomal dominant retinitis pigmentosa type 33; LMAN2L (OMIM #609552), associated with autosomal recessive mental retardation-52; and CNNM4 (OMIM #607805), related to autosomal recessive cone–rod dystrophy and amelogenesis imperfecta (Jalili syndrome, MIM #217080). ISCA search ((http://dbsearch.clinicalgenome.org/search/, April 2017) yielded nine similarly sized gains of copy variants, all categorized as “uncertain significance”. According to Clingen (June 2017), a score of 1 is given for triplosensitivity. Thus, the variant in case 3 was also defined as variants of unknown significance.
This study examined the frequency of chromosomal aberrations (both microscopic and submicroscopic) among cases of isolated kidney agenesis. It may be difficult to sometimes detect isolated cases, as minor fetal anomalies may be overlooked; some defects (i.e., patent ductus arteriosus) are a part of the normal fetal circulation and cannot be detected prenatally, and neurocognitive impairments cannot, obviously, be assessed by prenatal ultrasound. Therefore, some prenatal “isolated” cases may be actually syndromatic. This could be the case of the fetus diagnosed with isolated kidney agenesis and 22q11 microdeletion syndrome (DiGeorge syndrome). Nevertheless, this further emphasizes the fact that isolated unilateral kidney agenesis is not obviously associated with chromosomal aberrations, as CMA abnormalities are frequent in cases of multiple congenital anomalies, which may lead to an increased rate of abnormal CMA detection among fetuses with apparently isolated defects (24). However, the rate of chromosomal aberrations among fetuses with apparently isolated unilateral kidney agenesis was comparable with the results among low-risk pregnancies.
Our study has several prominent limitations. The first is the retrospective pattern of data acquisition, restricting the analysis to limited reported parameters. Unfortunately, we had no access to additional important factors, such as subsequent prenatal and postnatal follow-up. In addition, the precision of the final RR is poor, making any conclusions about the role of CMA in pregnancies with isolated unilateral renal agenesis unreliable. Finally, the level of the ultrasound evaluation was not included in the Ministry of Health database.
To the best of our knowledge, this paper is the first report describing the results of CMA testing in pregnancies with isolated unilateral renal agenesis. Although the two detected microdeletion syndromes found in our study are clearly pathogenic, and one of them is associated with high frequency or renal anomalies, the overall rate of chromosomal aberration detected is not different from that among fetuses with no sonographic abnormalities. Moreover, the rate of chromosomal aberrations detected in unilateral renal agenesis is much lower than its overall frequency in CAKUT (7).
Overall, it seems that pregnancies with isolated unilateral renal agenesis do not have an increased risk for abnormal CMA findings compared with fetuses with no evidence of congenital anomalies. Nevertheless, detection of two pathogenic CMA findings in our cohort, and the fact that in 1.03–1.39% of normal fetuses clinically significant CNVs are revealed incidentally, implies that CMA analysis in pregnancies with unilateral renal agenesis might still be useful, to the same degree as it can be in the general population. It seems that prenatal microarray analysis is more useful for the purpose of distinguishing among isolated and syndromic conditions, rather than identifying the genomic cause of the renal defect. A larger cohort of fetuses with isolated unilateral renal agenesis could shed more light on the indication for CMA testing in these pregnancies.
References
Natarajan G, Jeyachandran D, Subramaniyan B et al. Congenital anomalies of kidney and hand: a review. Clin Kidney J 2013;6:144–9.
Yosypiv IV . Congenital anomalies of the kidney and urinary tract: a genetic disorder? Int J Nephrol 2012;2012:909083.
Woolf AS, Hillman KA . Unilateral renal agenesis and the congenital solitary functioning kidney: developmental, genetic and clinical perspectives. BJU Int 2007;99:17–21.
Chow JS, Benson CB, Lebowitz RL . The clinical significance of an empty renal fossa on prenatal sonography. J Ultrasound Med 2005;24:1049–54 quiz 55-7.
Mesrobian HG, Rushton HG, Bulas D . Unilateral renal agenesis may result from in utero regression of multicystic renal dysplasia. J Urol 1993;150 (2 Pt 2): 793–4.
Cascio S, Paran S, Puri P . Associated urological anomalies in children with unilateral renal agenesis. J Urol 1999;162 (3 Pt 2): 1081–3.
Capone VP, Morello W, Taroni F et al. Genetics of congenital anomalies of the kidney and urinary tract: the current state of play. Int J Mol Sci 2017;18:4.
Uy N, Reidy K . Developmental genetics and congenital anomalies of the kidney and urinary tract. J PediatrGenet 2016;5:51–60.
Pallotta R, Bucci I, Celentano C et al. The 'skipped generation' phenomenon in a family with renal agenesis. Ultrasound Obstetr Gynecol 2004;24:586–7.
Nicolaides KH, Cheng HH, Abbas A et al. Fetal renal defects: associated malformations and chromosomal defects. Fetal Diagn Ther 1992;7:1–11.
Westland R, Verbitsky M, Vukojevic K et al. Copy number variation analysis identifies novel CAKUT candidate genes in children with a solitary functioning kidney. Kidney Int 2015;88:1402–10.
Kearney HM, Thorland EC, Brown KK et al. Working Group of the American College of Medical Genetics Laboratory Quality Assurance C. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med 2011;13:680–5.
South ST, Lee C, Lamb AN et al. ACMG Standards and Guidelines for constitutional cytogenomic microarray analysis, including postnatal and prenatal applications: revision 2013. Genet Med 2013;15:901–9.
Callaway JL, Shaffer LG, Chitty LS et al. The clinical utility of microarray technologies applied to prenatal cytogenetics in the presence of a normal conventional karyotype: a review of the literature. Prenatal Diagn 2013;33:1119–23.
Barber JC, Hall V, Maloney VK et al. 16p11.2-p12.2 duplication syndrome; a genomic condition differentiated from euchromatic variation of 16p11.2. Eur J Hum Genet 2013;21:182–9.
Hernando C, Plaja A, Rigola MA et al. Comparative genomic hybridisation shows a partial de novo deletion 16p11.2 in a neonate with multiple congenital malformations. J Med Genet 2002;39:E24.
Okamoto N, Fujii T, Tanaka J et al. A clinical study of patients with pericentromeric deletion and duplication within 16p12.2-p11.2. Am J Med Genet Part A 2014;164A:213–9.
McDonald-McGinn DM EB, Zackai EH. 22q11.2 deletion syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews®. Seatle, WA: University of Washington, Seattle. 1999:1993–2017.
Grati FR, Molina Gomes D, Ferreira JC et al. Prevalence of recurrent pathogenic microdeletions and microduplications in over 9500 pregnancies. Prenatal Diagn 2015;35:801–9.
McDonald-McGinn DM, Sullivan KE, Marino B et al. 22q11.2 deletion syndrome. Nat Rev Dis Primers 2015;1:15071.
Stewart TL, Irons MB, Cowan JM et al. Increased incidence of renal anomalies in patients with chromosome 22q11 microdeletion. Teratology 1999;59:20–2.
Besseau-Ayasse J, Violle-Poirsier C, Bazin A et al. A French collaborative survey of 272 fetuses with 22q11.2 deletion: ultrasound findings, fetal autopsies and pregnancy outcomes. Prenatal Diagn 2014;34:424–30.
Wu HY, Rusnack SL, Bellah RD et al. Genitourinary malformations in chromosome 22q11.2 deletion. J Urol 2002;168:2564–5.
Hillman SC, Pretlove S, Coomarasamy A et al. Additional information from array comparative genomic hybridization technology over conventional karyotyping in prenatal diagnosis: a systematic review and meta-analysis. Ultrasound Obstetr Gynecol 2011;37:6–14.
Acknowledgements
We are grateful to all Israeli genetic counselors and laboratory workers engaged in prenatal evaluation of fetal malformations, who made this study possible.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Sagi-Dain, L., Maya, I., Peleg, A. et al. Microarray analysis in pregnancies with isolated unilateral kidney agenesis. Pediatr Res 83, 825–828 (2018). https://doi.org/10.1038/pr.2018.3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2018.3
This article is cited by
-
Is an analysis of copy number variants necessary for various types of kidney ultrasound anomalies in fetuses?
Molecular Cytogenetics (2019)
-
Congenital Anomalies of the Kidney and Urinary Tract: a Clinical Review
Current Treatment Options in Pediatrics (2019)
