
Abstract
Alcohol use disorder (AUD) is a chronic debilitating disorder with limited treatment options and poorly defined pathophysiology. There are substantial genetic and epigenetic components; however, the underlying mechanisms contributing to AUD remain largely unknown. We conducted the largest DNA methylation epigenome-wide association study (EWAS) analyses currently available for AUD (total N = 625) and employed a top hit replication (N = 4798) using a cross-tissue/cross-phenotypic approach with the goal of identifying novel epigenetic targets relevant to AUD. Results show that a network of differentially methylated regions in glucocorticoid signaling and inflammation-related genes were associated with alcohol use behaviors. A top probe consistently associated across all cohorts was located in the long non-coding RNA growth arrest specific five gene (GAS5) (p < 10−24). GAS5 has been implicated in regulating transcriptional activity of the glucocorticoid receptor and has multiple functions related to apoptosis, immune function and various cancers. Endophenotypic analyses using peripheral cortisol levels and neuroimaging paradigms showed that methylomic variation in GAS5 network-related probes were associated with stress phenotypes. Postmortem brain analyses documented increased GAS5 expression in the amygdala of individuals with AUD. Our data suggest that alcohol use is associated with differential methylation in the glucocorticoid system that might influence stress and inflammatory reactivity and subsequently risk for AUD.
Similar content being viewed by others


Introduction
Multiple pathways to the development of alcohol use disorder (AUD) exist and include a complex interplay of environmental and genetic risk factors [1]. Genetic factors have been suggested to play a significant role in the etiology of AUD, as evidenced by twin, family, and adoption studies with heritability estimates ranging between 40 and 60% [2,3,4]; however, identifying the risk alleles has been difficult due to the complex mode of inheritance, significant clinical and genetic heterogeneity, and large number of genetic variants involved, each only contributing a small fraction to the overall risk.
The field of epigenetics is rapidly developing in AUD and might help explain some of the environmental components as they interact with the genetic architecture [5,6,7,8]. Several mechanisms contribute to epigenetic regulation, broadly defined as changes in gene expression without DNA sequence alterations, including histone modifications, non-coding RNA, and DNA methylation changes [9]. It is thought that various epigenetic mechanisms contribute to the pathophysiology of addictions. Some are drug-specific, while others are more generally involved in common pathways that lead to maladaptive and addictive behaviors [1, 10, 11].
While there has been some work on all of these epigenetic mechanisms in AUD, in particular, using various animal models [6, 12, 13], most studies were candidate gene driven and only a few studies used human tissue e.g. ref. [14]. Recent availability of DNA methylation array capture for comprehensive genome-wide profiling, has made it possible to conduct epigenome-wide association studies (EWASs). Only a few EWAS for AUD exist, but they are limited by small sample sizes, low array-capture, tissue types, inconsistent analysis strategy, and data interpretation [15,16,17,18,19,20,21,22,23,24,25]. Consequently, no universal DNA methylation loci for AUD have been identified; however, recent data suggest multiple loci associated with mild-moderate alcohol consumption [26] and interesting new targets for AUD [27].
To address these gaps in the literature, we conducted the largest EWAS analyses currently available for AUD using a cross-tissue/cross-phenotypic approach with the goal of identifying novel epigenetic targets relevant to AUD.
Materials and methods
Subjects
We used epigenome-wide data from six independent cohorts as follows (see also Fig. 1).

Six independent cohorts were assessed. These included the discovery sample, three replication cohorts, and two post-mortem brain cohorts. AUD alcohol use disorder, FDR false discovery rate, GS Generation Scotland: Scottish Family Health Study, GTP Grady Trauma Project, WCGNA weighted genome coregulation network analysis, BOLD blood oxygen level-dependent, fMRI functional magnetic resonance imaging, MRI magnetic resonance imaging, GAS5 growth arrest specific 5, FKBP5 FK506-binding protein 5.
Discovery cohort (blood, N = 539)
The discovery cohort comprised 539 participants (336 AUD and 203 controls) and was recruited at the National Institute on Alcohol Abuse and Alcoholism (NIAAA) at the National Institutes of Health (NIH), USA. All participants completed the Structured Clinical Interview for Diagnostic and Statistical Manual of Mental Disorders (DSM)-IV-TR (SCID-IV) [28] and an alcohol dependence (AD) diagnosis was determined. A diagnosis of AD in the DSM-IV is equivalent to moderate to severe AUD diagnosis according to the DSM-5 with a concordance of 93% [29]. In this manuscript we use AUD to be consistent with current nomenclature. Subjects completed several self-report questionnaires and clinical assessments. Peripheral blood was obtained for subsequent biomarker and DNA methylation analyses. All participants provided written informed consent in accordance with the Declaration of Helsinki and the study was approved by the Institutional Review Board of the NIAAA. Detailed demographic information can be found in Supplementary Table S1.
First replication cohort (blood, N = 86)
The first blood replication cohort included 86 participants (43 AUD and 43 controls) recruited to the NIAAA intramural program for a study on fear conditioning and extinction in AUD. Participants were included if they met the following criteria: between 21 and 65 years of age, able to provide written informed consent and cleared for venous access. In the AUD group, participants were also required to have a diagnosis of AD as assessed by the SCID-IV, to specify alcohol as their drug of choice, and to report alcohol consumption within the last 30 days on the Timeline Follow Back (TLFB) [30]. Participants were excluded if in their history and physical examination they reported neurological symptoms of the wrist or arm or reported chronic use of psychotropic medications within 4 weeks, fluoxetine use within 6 weeks, or incidental use of psychotropic medication within 5 half-lives of the beginning of the study. Additional exclusion criteria included presence of ferromagnetic implants, pregnancy, breastfeeding, left-handedness, claustrophobia, magnetic resonance imaging- (MRI-) incompatible intrauterine device or DSM-IV diagnosis of bipolar disorder, psychotic disorder, or current substance dependence other than alcohol, nicotine, or caffeine. Participants not seeking treatment for AUD were excluded if they had a history of alcohol-related seizures or presented with alcohol withdrawal symptom scores ≥ 8 on the Clinical Institute Withdrawal Assessment Alcohol Revised (CIWA-Ar) [31]. All participants provided peripheral blood for various biomarker assessments and DNA methylation studies. All participants provided written informed consent in accordance with the Declaration of Helsinki and the study was approved by the Institutional Review Board of the NIAAA. Detailed demographic information can be found in Supplementary Table S2.
Second replication cohort (blood, N = 4301)
The second replication cohort was from the Generation Scotland: Scottish Family Health Study (GS), a family-based cohort described elsewhere [32, 33]. Briefly, this cohort includes over 24,000 participants age 18–99 recruited between 2006 and 2011 for no specific disorder across Scotland. Alcohol consumption was assessed at baseline using a pre-clinical questionnaire and participants self-identified as current, former, or never drinkers. Average consumption was a self-report measure reflecting average weekly use in units. A table containing the units of alcohol contained in various drink types was available in order for participants to accurately estimate intake. All components of the GS received ethical approval from the National Health Service Tayside Committee on Medical Research Ethics (REC Reference Number: 05/S1401/89) and written consent was obtained from all participants. DNA methylation data was obtained for 5190 GS individuals from peripheral blood samples taken at baseline using the Infinium MethylationEPIC BeadChip. After quality control, 4301 participants were included in the analyses.
Third replication cohort (blood, N = 391)
The third blood replication cohort included participants from the Grady Trauma Project (GTP), and details of the sample have been described previously [34,35,36]. In brief, the GTP was a study on stressful life events and their predictors in a predominantly African American, urban population of low socioeconomic status. The sample included 1561 individuals who were recruited from a primary care clinic where they provided written informed consent, after which they completed a verbal interview and a blood draw. From this cohort, epigenetic information was available for 391 participants (females = 115, males = 276). The SCID-IV was administered and information on lifetime AUD was available for 328 of the 391 participants.
Postmortem brain cohorts (N = 58 and N = 46)
Information on DNA methylation of neural tissues was available from two postmortem cohorts. First, postmortem frontal cortex fluorescence activated cell sorted (FACS) neuronal and glial tissues of 58 individuals with and without major depressive disorder were available for analysis as described previously [37]. Of these 58 individuals, 7 had alcohol use problems (defined retrospectively based on responses to clinical interviews). Second, DNA methylation data on postmortem samples from a cohort of 46 participants with and without alcohol use problems was available from the Gene Expression Omnibus (GSE49393). This dataset included samples from individuals with DSM-IV defined AD (n = 9), alcohol abuse (n = 14), as well as an age-matched healthy control group (n = 23). For our study, we combined data from participants with AD and alcohol abuse to define a new group, ‘AUD’ (n = 23). Sample collection and processing for this cohort have been described in a previous publication [38].
Methylomic profiling
DNA methylation data from whole blood samples were assessed using an Infinium MethylationEPIC BeadChip microarray (Illumina Inc., San Diego, California) according to the manufacturer’s protocol for the discovery, first, and second replication cohorts (NIAAA and GS). The third replication sample (GTP) and neural tissues from both postmortem cohorts were assessed with the Illumina 450K chip as described previously [37, 38]. Pre-processing of the GS data has been described in detail elsewhere [39].
Statistical analysis
Raw data from discovery and replication cohorts were processed with the package ‘wateRmelon’ in R [40]. All cross-reactive probes and probes failing quality assessment were removed. Scale-based correction was applied to Illumina type I versus type II probes. Methylated and unmethylated intensities were quantile-normalized in the red and green channels separately using the Dasen method in WateRmelon [40], followed by β-value (intensity ratios of methylated to unmethylated probes) calculation. All analyses were carried out in R, version 3.5.1 (© 2018 The R Foundation for Statistical Computing).
Linear regressions were used to examine the associations of each CpG site with AUD in the discovery and first replication cohort. DNA methylation beta values were regressed on AUD status adjusting for age, sex, race, and cell type composition of blood. Cell type composition was derived from DNA methylation proxies using the Houseman algorithm [41]. Significant probes (p ≤ 0.05) in both the discovery and first replication cohorts were identified and further examined as follows.
All probes identified above were tested for associations with AUD diagnosis in the second (GS) and third (GTP) blood replication cohorts. Associations were tested in the GTP cohort using linear regressions as described above. In the GS cohort, linear regression models were fit in the limma R packages with CpG site as an outcome variable and log transformed units per week as an independent variable. Age, sex, smoking status, pack years of smoking, and the first 20 principal components from the M-values corrected for age, sex, relatedness batch, and estimated cell counts were fit as covariates in the linear model.
Postmortem brain analysis
Methylation levels of the target probes were regressed on alcohol use status controlling for age, sex, and race (only in the first postmortem set), either separately for neuronal and glial tissue (first postmortem dataset) or adjusting for neural cell type proportion (second postmortem dataset).
Weighted genome coregulation network analysis (WGCNA)
WGCNA was performed to detect clusters (modules) of highly correlated genes using the top 96 probes exhibiting consistent FDR significance with AUD diagnosis from the discovery and replication cohorts. Module detection was carried out using a soft thresholding power β of 9, which was chosen by maximizing scale free topology model fitting as a function of model connectivity based on the internal data structure according to the recommendations of Horvath [42]. All analyses were performed in R.
Mediation network analysis
A mediation analysis approach was performed on WGCNA identified module loci such that each loci in the top 30th percentile of module membership or ‘hubness’ is assessed for association to the outcome metric (cortisol, see results), both alone and in conjunction with every other module locus to assess for mediation. Those connections implicated as mediating by means of statistical association below 5% with the outcome alone and no association in an additive model are considered ‘connected’. Connected genes are quantified and displayed using the iGraph package in R.
Endophenotype analysis
Cortisol measurements and assessments
All blood samples were taken before 09:00 AM after inpatient admission. Cortisol was measured using radioimmunoassay (RIA) with an intra-assay and inter-assay coefficient of variation of 3.5% and 14.3%, respectively.
BOLD fMRI of fear conditioning and extinction (brain; N = 86)
Subject description provided under first replication cohort.
Fear conditioning task: The Fear Conditioning and Extinction (FCE) experiment took place after individuals with AUD had no withdrawal symptoms for at least two consecutive days. Day 1 consisted of habituation, conditioning, and two extinction blocks and Day 2 consisted of extinction recall and renewal blocks. Galvanic skin responses were obtained throughout the fMRI session using Ag/AgCl electrodes (41 mm diameter). Two additional electrodes were placed on the wrist of the same hand to deliver electrical stimulation (unconditioned stimulus [US]; 0.5 s duration). The intensity level of electrical stimulation was uncomfortable but tolerable as determined by a personalized work-up on the first day. Participants were presented with digital photographs of two different rooms, each containing a lamp shade that turned blue or yellow as the conditioned stimuli (CSs). During conditioning, one of the colors was paired with a 0.5-s electrical stimulation (CS+) in 75% of the trials, while the other was never followed by a shock (CS−). For further details please see [43].
fMRI data processing: Functional data were processed using Statistical Parametric Mapping (SPM12b, Wellcome Department of Cognitive Neurology, London, UK) based on MATLAB R2018b (MathWorks, Natick, MA, USA). After removal of the first three individual functional scans of the experimental conditioning phase to avoid artefacts caused by magnetic saturation effects, and prior to preprocessing, all images were visually controlled for gross movement artefacts and anatomical abnormalities; all images of the 86 subjects were included. Scans were further corrected for signal‐to‐noise decrease in single slices, using the denoising function of the ArtRepair software (http://cibsr.stanford.edu/tools/human-brain-project/artrepair-software.html, assessed 03 Jan, 2019). Afterwards, individual scans were spatially realigned to correct for head motion and normalized using the warping parameters estimates of the individual co-registered and segmented MPRGE image. Images were normalized to an isovoxel size of 3.5 × 3.5 × 3.5 mm. Subsequent smoothing was done using an isotropic Gaussian kernel (8 mm FWHM).
The preprocessed fMRI data were then analyzed as a block design for the conditioning phase in the context of the general linear model approach using a two‐level procedure. On the individual single subject level, the different conditions (CS+ and CS−) were modeled (boxcar functions convolved with the hemodynamic response function) as explanatory variables together with the six movement parameters to account for residual variance due to head motion, and a single constant representing the mean over scans. Both CS+ and CS− conditions in the fear conditioning phase were modeled as separate regressors for trials 2–20 since learning of CS+ (potential shock) versus CS- (no shock) has not occurred yet during the first trial. Subsequently, for each subject, linear contrast images were computed for fear conditioning: ‘CS+ minus CS−‘, trials 2–20. For these individual contrast images, overall blood oxygenation level-dependent (BOLD) responses were assessed for the “effect of interest” provided by an analysis of variance. Here, we specifically assessed small volume adjusted BOLD responses of anatomical atlas-based a priori Regions of Interest (ROI) as brain regions crucially involved in emotion detection and regulation, i.e., left/right amygdala, left/right hippocampus, left/right medial prefrontal cortex (mPFC) (i.e., aal-mask for Front_Med_Orb), left/right insula, left/right rostral anterior cingulate cortex (rACC, ROI as defined in a prior study [44], using the WFU PickAtlas toolbox (http://fmri.wfubmc.edu/software/PickAtlas). Parameter estimates of the ROI BOLD responses (identifying clusters with an initial voxel-level threshold of P (uncorrected) < 0.005) were then extracted from the cluster peak as SPM eigenvariates for further BOLD analyses as described below.
BOLD analysis: Associations between blood methylation levels for our target probes and BOLD signal data (from the first replication cohort) were examined. As the normality assumption for residuals was not met for regressions of methylation on BOLD signal, we used non-parametric Kendall’s partial correlations. For this, we regressed out inter-individual differences in cell type composition and estimated the correlations of the residuals with BOLD signal, adjusting for age, sex, and race.
Structural MRI (brain; N = 193)
A subset of participants from the discovery cohort also participated in a structural MRI study. Hippocampus volumes were determined using the standard Freesurfer [45] (version 5.3.0; surfer.nmr.mgh.harvard.edu) pipeline. The individual T1-weighted images were automatically segmented to measure gray matter volume of structures [46] using the following steps: (1) The images were resampled to 1 mm3 voxels; transformed to Talairach space, the intensity non-uniformity corrected [47]; and skull was stripped from the images [48]; and finally auto-segmentation proceeded with labels assigned based on probabilistic location of structures. We conducted a reliability test by examining a random number of the auto-segmented volumes with recon_checker from FreeSurfer’s QATools. This included checking for outliers, calculating signal-to-noise ratio, and visually examining generated snapshots of brain volume segmentation.
Targeted human postmortem brain mRNA analyses (brain; N = 24)
Postmortem tissues were obtained from the New South Wales Tissue Resource Centre (NSWBTRC) at the University of Sydney, Australia. Brain tissues from 11 males with AUD and 13 controls were analyzed for amygdala and prefrontal cortex (PFC). All AUD subjects had alcohol detected in blood and were also daily smokers at the time of death. Total RNA was extracted from male postmortem frozen brain tissue, using the RNeasy Lipid Tissue mini Kit (Qiagen). One µg total RNA was reverse-transcribed using SuperScript® III First-Strand Synthesis SuperMix for qRT-PCR (Invitrogen). Real-time quantitative polymerase chain reaction (PCR) was run in ViiA™ 7 Real-Time PCR System using TaqMan Gene Expression Assays (GAS5; Hs03464472_m1, Thermo Fisher). Data and statistical analyses were performed using GraphPad prism 8.0 (GraphPad Software Inc., San Diego, CA, USA). An unpaired t-test was used to determine statistical significance with p < 0.05.
Results
Associations of DNA methylation in blood with AUD status
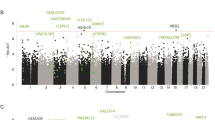
We identified 69242 CpG probes that were significantly (p ≤ 0.05) associated with AUD in the discovery cohort and 72941 probes in the first replication cohort, using a linear regression model additively controlling for age, sex, and race as covariates. Demographic and sample characteristics can be found in Supplementary Tables S1 and S2. After false discovery rate (FDR) correction for multiple testing (FDR threshold p < 0.05), 5101 probes in the discovery and 203 probes in the first replication sample remained significant (Figs. 1, 2a, b). Of these, 96 probes were consistently FDR significant in both cohorts (notably all in the same direction), representing an overlap significantly higher than that expected by chance (expected probability= 0.0006; 95% CI = 0.40–0.54) (Supplementary Table S3). To further refine this set, we performed additional replication analyses using linear models for quantitative alcohol consumption in Generation Scotland: the Scottish Family Health Study (GS:SFHS) and SCID lifetime AUD diagnosis in the Trauma Project (GTP) (see Supplementary Tables S4, S5). Of the 96 originally identified loci, a total of 70 exhibited significant (p < 0.05) associations in at least one of the two cohorts. Notably, two probes located within the long non-coding RNA growth arrest specific 5 gene (GAS5) (GAS5a = cg06644515, GAS5b = cg16290996) were among the CpGs most consistently associated with AUD or alcohol use across all cohorts investigated (Table 1).

Volcano plots depicting the effect sizes of DNA methylation association with AUD (x axis) as a function of the negative natural log of the p value (y axis) for the discovery (a) and replication (b) cohorts. Red dashed horizontal line depicts a nominal p value of 0.05. c Scatter plot showing the relationship between module membership of probes (x axis) versus associations with AUD status (y axis).
As smoking is commonly comorbid with AUD and also leads to DNA methylation changes, we carried out additional regressions to test if adjusting for smoking altered our results for the 96 target probes. Methylation at each of the 96 probes was regressed on AUD status, controlling for smoking (self-reported scores on the Fagerstrom’s test for nicotine dependence) in addition to age, sex, race, and cell type. All probes retained significant associations after adjusting for smoking scores (Supplementary Table S6).
In order to address possible dose/life-time alcohol exposure on methylation, we carried out additional exploratory analyses in the discovery cohort. A metric of exposure was calculated by multiplying the average number of drinks per day by the number of years since their self-reported age of drinking inception. Of the 96 CpG sites, 65 probes exhibited significant associations with dose (Supplementary Table S7), suggesting that the observed epigenetic associations in these cases may be a consequence of alcohol exposure. Subset analyses for DNA methylation changes by ethnicity and gender are shown in Supplementary Fig. 1 and Supplementary Table S8.
Weighted genome coregulation network analysis
Given that methylomic variation is often correlated across CpG sites, we conducted a Weighted Gene Coregulation Network Analysis (WGCNA) adjusting for age, sex, and ethnicity in the 96 probes significantly associated in both the discovery and replication cohorts. We identified one significant AUD associated module consisting of 50 probes (Module association Rho= 0.76, P = 2.2 × 10−16, Supplementary Table S9), containing glucocorticoid signaling associated genes GAS5b, and FKBP5, inflammatory cytokine driving genes LURAP1L and LURAP1L-AS1, and a number of small nucleolar RNA (snoRNA) targets, among others.
Gene ontology analysis
A gene ontology analysis identified a number of significant biological pathways, including some potentially indicative of dysregulated inflammatory processes (Supplementary Table S10).
Association of top AUD associated loci and coregulated modules with cortisol
Based on the WCGNA analyses which revealed top targets relevant to the glucocorticoid system, we investigated DNA methylation associations with morning cortisol (Fig. 3a). Morning cortisol levels collected under the same protocol were available across the discovery and first replication cohorts and were subsequently combined and assessed for associations with AUD coregulated module variation. The first eigenvector of a principal component analysis was generated for those 50 loci in the significant AUD associated module identified by WGCNA in order to represent the majority of this coregulated epigenetic variation. A significant association with morning cortisol levels was observed after adjusting for age, sex, and ethnicity (b = −3.85 ± 1.63, F = 5.14, df= 4/309, p = 0.019) (Fig. 3). To assess module gene functional importance to cortisol variation, we employed a mediation network analysis approach to identify loci with the highest number of cortisol associated connections among coregulated genes. Of those 50 loci interrogated, GAS5b exhibited the most evidence for connectedness in association to cortisol (N = 18) (Fig. 3b). In light of its functional relationship with HPA axis regulation, we assessed the association between two of the GAS5 probes and observed a significant association with GAS5b (b = ‒17.87 ± 8.62, F = 4.70, df = 4/309, p = 0.039) but not GAS5a (data not shown) (Fig. 3b).

a Scatterplot of morning cortisol (y axis) as a function of AUD associated coregulated module eigenvector values (x axis). b Network diagram from the network mediation analysis demonstrating coregulated genes associated with morning cortisol in the discovery dataset. c Scatterplot of morning cortisol (y axis) as a function of GAS5b DNA methylation (x axis).
Association of DNA methylation with stress-related AUD neuroimaging endophenotypes
We assessed BOLD activation during fear acquisition to assess how DNA methylation associated with AUD was associated with BOLD signal in different brain regions. Significant associations (p ≤ 0.05) were observed after adjusting for age, sex, and race in the left amygdala, and both the left and right insula (Table 2 and Supplementary Table S10). We next assessed associations with morning cortisol and identified significant associations in both the left and right vmPFC, and the right ACC (Table 2). As the AUD associated coregulated DNA methylation module above was associated with cortisol, we investigated the association of the eigenvector with BOLD activation levels and observed significant associations in the right amygdala and a non-significant trend for association with the left vmPFC (Fig. 4a, b). Individual results for the original 96 probes are reported in Supplementary Table S10. Together, the results suggest that DNA methylation changes in response to alcohol exposure have the potential to mediate altered brain activity patterns through alteration of HPA axis activity and stress sensitivity.

a A scatterplot of amygdala BOLD activation (x axis) as a function of AUD associated module eigenvector variation (y axis) in the fear cohort. b A scatterplot of vmPFC BOLD activation (x axis) as a function of AUD associated module eigenvector variation (y axis) in the fear cohort. c A scatterplot of right hippocampal volume (x axis) as a function of AUD associated module eigenvector variation (y axis) in the discovery cohort.
Association of AUD DNA methylation module eigenvector with hippocampal structure
In light of the observed AUD associations with epigenetic regulation of cortisol signaling and previous work demonstrating associations of cortisol with limbic cortical volume [49], we investigated available structural measures with AUD associated epigenetic variation. In the discovery sample, the AUD associated coregulated module eigenvector was significantly associated with right, but not left hippocampal volume after adjusting for age, sex, and race (right β = 546.55 ± 200.021, F = 7.103, df = 5/177, p = 0.0069; left β = 395.528 ± 219.9, F = 3.97, df = 5/177, p = 0.074) (Fig. 4c).
Association of AUD and DNA methylation in postmortem tissue
Both PRKCZ and GAS5 were consistently identified in two independent brain tissue cohorts, suggesting these may be among the most robust brain related findings identified. Of the 96 target probes identified in the discovery and first blood replication datasets, only 27 were available for examination in the NICHD postmortem brain dataset (due to differences in the array platforms EPIC 850K versus 450K). In FACS isolated glial tissue, alcohol use status (N = 29 cases, N = 29 controls) was associated with PRKCZ methylation (Supplementary Table S11). In neuronal tissue, alcohol use status (N = 29 cases, N = 29 controls) was associated with methylation levels of GAS5, GLTSCR1, and B2M (Supplementary Table S11). Methylation data were available for 34 of the 96 target probes in the Australian Brain Bank postmortem brain dataset (N = 23 cases, N = 23 controls) (GSE49393). DNA methylation of probes located within UBA3, CABLES1, MYST3, GAS5, MYH10, TOLLIP, HNRNPA-1, PRKCZ, and GLTSCR1 was significantly associated with AUD status (Supplementary Table S11).
Expression analysis of GAS5 in human postmortem brain
Given the consistent association of GAS5 methylation with AUD phenotypes, we analyzed expression of GAS5 in 11 individuals with AUD and 13 controls using human postmortem brain tissues including amygdala and prefrontal cortex. Results showed statistically significantly increased GAS5 expression in the amygdala in AUD (p < 0.05) (Fig. 5), indicating a potential effect of alcohol on GAS5 expression.

a mRNA expression in human amygdala. b mRNA expression in human PFC. Controls n = 13, case n = 11. *p < 0.05. AMY = amygdala, PFC = prefrontal cortex.
Discussion
This work represents the largest and most comprehensive EWAS on AUD to date. The large number of significant probes identified in our study may derive both from the statistical power inherent in large cohorts as well as widespread epigenetic changes induced by chronic alcohol exposure. In our discovery cohort, we identified over 5000 DNA methylation associations with AUD after correction for multiple testing. Importantly, while many of these associations are likely biologically relevant, we sought a means beyond statistical stratification to identify the strongest findings from this set. Cross referencing discovery findings with those derived from independent replication sets represents a powerful tool to identify robust findings with likely biological meaning. Using this method, we identified 96 loci consistently associated with both our initially discovered ~5000 FDR significant probes and the 203 FDR significant probes in our first replication set. The overlap between these cohorts was much higher than expected by chance as was an enrichment of multiple biological pathways exhibiting epigenetic change. We further attempted to replicate our findings in a second and third independent population-based cohort. Assessment of the GS cohort revealed 70 of the 96 probes to be significantly associated with alcohol use. The GTP data was generated on an earlier technology, the HM450 microarray, and contained only 35 of the 96 loci above, of which 15 loci were identified to be associated with SCID based AUD diagnoses. Genes associated with these probes included GAS5, MYST3, UBA3, HECW2, MYH10, RASGRP1, PRKCZ, and FKBP5. Gene ontology (GO) analysis demonstrated strong evidence for AUD associated epigenetic alterations in genes associated with immune response, glucocorticoid signaling and various inflammatory cytokines (Supplementary Table S12). Notably, the effects of alcohol exposure on inflammatory processes are well documented. Alcohol exposure can activate both acute and chronic inflammatory processes, both in brain [50] and liver [51].
Given that methylomic variation across the genome is not independent and likely coregulated by various systematic influences, we sought to further disentangle and model the biological complexity of identified CpG sites in our sample. We used WGCNA to generate a data driven grouping of loci most likely to be coregulated in association with AUD. One module consisting of 50 loci was associated with AUD status and contained a number of loci relevant to alcohol exposure (Result S1), including glucocorticoid signaling and inflammation. Interestingly, our data are in line and partially replicate a recent study of DNA methylation in human postmortem brain, which identified probes in the glucocorticoid receptor and FKBP5 in AUD [22].
Based on these promising findings, both from a single CpG standpoint (robust findings for GAS5 across all datasets) and the WGCNA analyses, we conducted several follow up experiments to investigate biological function with relevance to human stress responsiveness.
First, we established that the AUD WGCNA module was associated with morning cortisol levels. Remarkably, of those 50 loci interrogated, a network mediation analysis suggested that a probe in GAS5 exhibited the highest evidence for connectedness in association to cortisol regulation by mediating the association with cortisol across a large number of module genes. GAS5 interacts with the glucocorticoid receptor (GR) to inhibit its transcriptional function [52] by folding into a soluble glucocorticoid response element-like sequence on the GR to mimic GR binding [53]. Furthermore GAS5 expression may also alter corticotrophin releasing hormone receptor 1 (CRHR1) expression [54]. This finding is intriguing, given robust literature supporting an involvement of the corticotropin releasing factor and its receptor in AUD [55,56,57,58,59,60].
Second, we used neuroimaging paradigms to probe key regions implicated in stress responsiveness in human, to further explore biological mechanisms of identified CpG-related networks. Main brain regions implicated in the human stress response include the amygdala, frontal cortex, and hippocampus, among others [61, 62]. Importantly, we aimed to explore the neuronal and functional relevance of peripherally identified epigenetic changes. As demonstrated by our group previously, such peripherally identified epigenetic effects may have brain relevance when they derive from a systemic factor such as circulating steroid hormones [63] or alcohol [27]. We focused mainly on the frontal cortex, hippocampus and amygdala. To probe the amygdala specifically, we used a fear conditioning and extinction paradigm in individuals with AUD and controls. Our data showed robust association of AUD-associated DNA methylation with amygdala activation to the anticipation of electric shocks in humans. These data suggest that the AUD-associated coregulated module genes are associated with BOLD signals in distinct brain regions, which in turn may reflect brain-related phenotypes (Supplementary Table S10, Result S1). Remarkably, AUD module epigenetic variation was also associated with hippocampal volume, suggesting a further link between peripheral DNA methylation variation and brain phenotypes. Summarizing our endophenotypes studies, we observed that GAS5 DNA methylation was associated with alcohol use across all studied cohorts both in the periphery and in the brain. Furthermore, we studied GAS5 gene expression by conducting postmortem studies of AUD patients and controls and found higher GAS5 expression in the amygdala. This is intriguing, given the fMRI finding in the amygdala, it suggests a potential direct role of GAS5 in this brain region, while further corroborating the functional relevance of our results. The importance of GAS5 on phenotypic outcomes may be attributed to its implicated role as a ‘hub gene’ regulating glucocorticoid signaling. GAS5 exhibited strong module membership (hub status) and demonstrated evidence for a high degree of network connectedness in association with cortisol levels, suggesting it may play a central role in the observed association between AUD co-regulated module variation and cortisol status. It further suggests that BOLD-associated epigenetic variation may be a downstream consequence of or interact with dysregulated glucocorticoid signaling. These results are consistent with the known function of GAS5 and other module genes exhibiting evidence for coregulation such as FKBP5 as both are involved in modulating glucocorticoid signaling, and broadly, inflammation. As such, previous evidence linking FKBP5 to AUD may be in some way be related to GAS5 and should be investigated further. For example, FKBP5 is transcribed by the GR and, once expressed, represents a potent intracellular inhibitor of GR function [52]. FKBP5 gene expression has been suggested as an important mediator of the pathways to the development of drinking behavior. Previous studies show that genetic variants within FKBP5 that affect its expression mediate effects of both poor parental relationships [64] and metacognitions about alcohol on problematic drinking behavior [65]. These findings hint at genetic effects that modulate FKBP5 expression, which may contribute to AUD. Similar to our results, a recent study showed methylation changes in FKBP5 in the prefrontal cortices of adults with alcohol use problems [22]. Concurrently, animal studies demonstrate that FKBP5 expression in mesocorticolimbic dopaminergic neurons mediates the effects of early life stress on alcoholic behavior [64]. While we observed epigenetic associations with FKBP5 in three cohorts (discovery and replication AUD, GTP), we could not detect such an effect in the GS sample, possibly due to variation in the degree of trauma/early life stress exposure. Moreover, epigenetic variation in FKBP5 is particularly interesting in the context of fear conditioning, as was tested in our study participants. FKBP5 may confer risk to fear extinction deficits [66], while, epigenetic regulation of FKBP5 has been proposed as a mechanism to mediate glucocorticoid exposure enhanced fear extinction in rodents [67].
Despite our promising findings, there are some limitations that should be carefully considered. While we used several cohorts to replicate methylomic variation associated with AUD, it is important to keep in mind that other factors besides alcohol might influence epigenetic signatures, such as clinical heterogeneity and environmental factors including life-experiences, trauma, diet, exercise patterns, and underlying genetic architecture. In addition, the mechanisms by which alcohol leads to widespread epigenetic reprogramming are not addressed by our study. One possibility is that epigenetic change is the downstream consequence of the secondary effects of alcohol exposure such as inflammatory changes or disrupted glucocorticoid signaling. Alternatively, other loci may represent more direct mediators of the effects of alcohol on phenotype or etiological factors leading to the AUD phenotype. For example, acute alcohol consumption can lead to brain expression changes in PRKCZ (one of the replicating genes identified in our study that was not a member of the AUD associated coregulated module), suggesting it may be a consequence of alcohol exposure. Alternatively, deletion of PRKCZ in mice leads to increased consumption of alcohol [68] suggesting that variation in PRKCZ precedes the AUD phenotype. Similarly, methylation at GAS5 could reflect a mediating effect of alcohol. Previous work has demonstrated that GAS5 is bound by the catalytic EZH2 subdomain of polycomb group complex 2 (PRC2) [69]. Further, a recent report highlighted a mechanism whereby alcohol exposure resulted in histone 3 lysine 27 trimethylation marks (within the amygdala) on a brain-derived neurotrophic factor associated long non-coding RNA by EZH2 [70]. Although the degree to which such a mechanism is generalizable for other non-coding RNAs like GAS5 has yet to be determined, it is possible that DNA methylation of GAS5 is affected by interaction with PRC2 and which reflects a mediating effect of alcohol via EZH2. As such, the translation from histone modifications to DNA methylation through sequestration of DNA methyltransferase activity at this specific locus will need to be determined through future studies. Importantly, understanding the cause vs. effect nature of the identified associations is not possible in a cross-sectional design such as the one applied in our study and calls for comprehensive longitudinal studies paired with preclinical animal work to better understand etiological mechanisms in humans. Despite this, consistent AUD associated findings across multiple cohorts suggest the possibility of identifying a combined ‘biosignature’ of AUD. A better understanding of the association of epigenetic variation with not only the diagnostic criteria itself, but also disease-associated endophenotypes such as brain imaging alterations may eventually aid in the development of objective clinical tools to assess for exposure to alcohol and the progression through various phases of drinking behavior severity.
A strength of this study is the use of both diagnostic and dimensional definitions of chronic alcohol use. Across cohorts with various definitions, we continued to find the same probes to be differentially methylated in response to alcohol use, suggesting that the probes identified here are particularly robust indicators of alcohol induced epigenetic changes.
References
Ron D, Barak S. Molecular mechanisms underlying alcohol-drinking behaviours. Nat Rev Neurosci. 2016;17:576–91.
Enoch MA, Goldman D. The genetics of alcoholism and alcohol abuse. Curr Psychiatry Rep. 2001;3:144–51.
Agrawal A, Lynskey MT. Are there genetic influences on addiction: evidence from family, adoption and twin studies. Addiction. 2008;103:1069–81.
Tawa EA, Hall SD, Lohoff FW. Overview of the genetics of alcohol use disorder. Alcohol Alcohol. 2016;51:507–14.
Schuebel K, Gitik M, Domschke K, Goldman D. Making sense of epigenetics. Int J Neuropsychopharmacol. 2016;19:pyw058.
Palmisano M, Pandey SC. Epigenetic mechanisms of alcoholism and stress-related disorders. Alcohol. 2017;60:7–18.
Luo A, Jung J, Longley M, Rosoff DB, Charlet K, Muench C, et al. Epigenetic aging is accelerated in alcohol use disorder and regulated by genetic variation in APOL2. Neuropsychopharmacol. 2020;45:327–36.
Charlet K, Rosenthal A, Lohoff FW, Heinz A, Beck A. Imaging resilience and recovery in alcohol dependence. Addiction. 2018;113:1933–1950.
Nestler EJ. Epigenetic mechanisms of drug addiction. Neuropharmacology. 2014;76 Pt B:259–68.
Robison AJ, Nestler EJ. Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci. 2011;12:623–37.
Kessler RC, Crum RM, Warner LA, Nelson CB, Schulenberg J, Anthony JC. Lifetime co-occurrence of DSM-III-R alcohol abuse and dependence with other psychiatric disorders in the National Comorbidity Survey. Arch Gen Psychiatry. 1997;54:313–21.
Starkman BG, Sakharkar AJ, Pandey SC. Epigenetics-beyond the genome in alcoholism. Alcohol Res: Curr Rev. 2012;34:293–305.
Berkel TD, Pandey SC. Emerging role of epigenetic mechanisms in alcohol addiction. Alcohol Clin Exp Res. 2017;41:666–80.
Hagerty SL, Bidwell L, Harlaar N, Hutchison KE. An exploratory association study of alcohol use disorder and DNA methylation. Alcohol: Clin Exp Res. 2016;40:1633–40.
Manzardo AM, Henkhaus RS, Butler MG. Global DNA promoter methylation in frontal cortex of alcoholics and controls. Gene. 2012;498:5–12.
Xu H, Wang F, Kranzler HR, Gelernter J, Zhang H. Alcohol and nicotine codependence-associated DNA methylation changes in promoter regions of addiction-related genes. Sci Rep. 2017;7:41816.
Xu H, Wang F, Liu Y, Yu Y, Gelernter J, Zhang H. Sex-biased methylome and transcriptome in human prefrontal cortex. Hum Mol Genet. 2013;23:1260–70.
Zhang H, Wang F, Kranzler HR, Zhao H, Gelernter J. Profiling of childhood adversity-associated DNA methylation changes in alcoholic patients and healthy controls. PLoS ONE. 2013;8:e65648.
Weng JT, Wu LS, Lee CS, Hsu PW, Cheng AT. Integrative epigenetic profiling analysis identifies DNA methylation changes associated with chronic alcohol consumption. Comput Biol Med. 2015;64:299–306.
Zhang R, Miao Q, Wang C, Zhao R, Li W, Haile CN, et al. Genome-wide DNA methylation analysis in alcohol dependence. Addiction Biol. 2013;18:392–403.
Zhao R, Zhang R, Li W, Liao Y, Tang J, Miao Q, et al. Genome-wide DNA methylation patterns in discordant sib pairs with alcohol dependence. Asia Pac Psychiatry. 2013;5:39–50.
Gatta E, Grayson DR, Auta J, Saudagar V, Dong E, Chen Y et al. Genome-wide methylation in alcohol use disorder subjects: implications for an epigenetic regulation of the cortico-limbic glucocorticoid receptors (NR3C1). Mol Psychiatry 2019. [Epub ahead of print]
Philibert R, Plume J, Gibbons F, Brody G, Beach S. The impact of recent alcohol use on genome wide DNA methylation signatures. Front Genetics 2012;3:54.
Philibert RA, Penaluna B, White T, Shires S, Gunter T, Liesveld J, et al. A pilot examination of the genome-wide DNA methylation signatures of subjects entering and exiting short-term alcohol dependence treatment programs. Epigenetics. 2014;9:1212–9.
Zhang H, Herman AI, Kranzler HR, Anton RF, Zhao H, Zheng W, et al. Array-based profiling of DNA methylation changes associated with alcohol dependence. Alcohol: Clin Exp Res. 2013;37:E108–E115.
Liu C, Marioni RE, Hedman AK, Pfeiffer L, Tsai PC, Reynolds LM, et al. A DNA methylation biomarker of alcohol consumption. Mol Psychiatry 2018;23:422–433.
Lohoff FW, Sorcher JL, Rosen AD, Mauro KL, Fanelli RR, Momenan R, et al. Methylomic profiling and replication implicates deregulation of PCSK9 in alcohol use disorder. Mol psychiatry. 2018;23:1–11.
First MB, Spitzer RL, Gibbon M, Williams JB. Structured clinical interview for DSM-IV axis I disorders. New York: New York State Psychiatric Institute; 1995.
Goldstein RB, Chou SP, Smith SM, Jung J, Zhang H, Saha TD, et al. Nosologic comparisons of DSM-IV and DSM-5 alcohol and drug use disorders: results from the national epidemiologic survey on alcohol and related conditions–III. J Stud Alcohol Drugs. 2015;76:378–88.
Sobell LC, Sobell MB. Timeline followback user’s guide: a calendar method for assessing alcohol and drug use. Toronto: Addiction Research Foundation Toronto; 1996.
Sullivan JT, Sykora K, Schneiderman J, Naranjo CA, Sellers EM. Assessment of alcohol withdrawal: the revised clinical institute withdrawal assessment for alcohol scale (CIWA-Ar). Br J Addict. 1989;84:1353–7.
Smith BH, Campbell H, Blackwood D, Connell J, Connor M, Deary IJ, et al. Generation Scotland: the Scottish Family Health Study; a new resource for researching genes and heritability. BMC Med Genet. 2006;7:74.
Smith BH, Campbell A, Linksted P, Fitzpatrick B, Jackson C, Kerr SM, et al. Cohort Profile: Generation Scotland: Scottish Family Health Study (GS:SFHS). The study, its participants and their potential for genetic research on health and illness. Int J Epidemiol. 2012;42:689–700.
Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB, et al. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA. 2008;299:1291–305.
Ressler KJ, Mercer KB, Bradley B, Jovanovic T, Mahan A, Kerley K, et al. Post-traumatic stress disorder is associated with PACAP and the PAC1. receptor 2011;470:492.
Gillespie CF, Bradley B, Mercer K, Smith AK, Conneely K, Gapen M, et al. Trauma exposure and stress-related disorders in inner city primary care patients. General Hospital Psychiatry. 2009;31:505-514.
Guintivano J, Aryee MJ, Kaminsky ZA. A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics. 2013;8:290–302.
Xu H, Wang F, Liu Y, Yu Y, Gelernter J, Zhang H. Sex-biased methylome and transcriptome in human prefrontal cortex. Hum Mol Genet. 2014;23:1260–70.
McCartney DL, Stevenson AJ, Walker RM, Gibson J, Morris SW, Campbell A, et al. Investigating the relationship between DNA methylation age acceleration and risk factors for Alzheimer’s disease. Alzheimers Dement (Amst). 2018;10:429–37.
Pidsley R, CC YW, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics. 2013;14:293.
Houseman EA, Kelsey KT, Wiencke JK, Marsit CJ. Cell-composition effects in the analysis of DNA methylation array data: a mathematical perspective. BMC Bioinforma. 2015;16:95.
Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:17.
Muench C, Charlet K, Balderston NL, Grillon C, Heilig M, Cortes CR et al. Fear conditioning and extinction in alcohol dependence: evidence for abnormal amygdala reactivity. Addiction Biol. 2019:e12835. [Epub ahead of print]
Charlet K, Schlagenhauf F, Richter A, Naundorf K, Dornhof L, Weinfurtner CE, et al. Neural activation during processing of aversive faces predicts treatment outcome in alcoholism. Addict Biol. 2014;19:439–51.
Fischl B. FreeSurfer. NeuroImage. 2012;62:774–81.
Destrieux C, Fischl B, Dale A, Halgren E. Automatic parcellation of human cortical gyri and sulci using standard anatomical nomenclature. NeuroImage. 2010;53:1–15.
Sled JG, Zijdenbos AP, Evans AC. A nonparametric method for automatic correction of intensity nonuniformity in MRI data. IEEE Trans Med Imaging. 1998;17:87–97.
Segonne F, Dale AM, Busa E, Glessner M, Salat D, Hahn HK, et al. A hybrid approach to the skull stripping problem in MRI. NeuroImage. 2004;22:1060–75.
Szeszko PR, Lehrner A, Yehuda R. Glucocorticoids and hippocampal structure and function in PTSD. Harv Rev Psychiatry. 2018;26:142–57.
Petrakis IL, Ralevski E, Gueorguieva R, Sloan ME, Devine L, Yoon G, et al. Targeting neuroinflammation with minocycline in heavy drinkers. Psychopharmacology. 2019; 236:3013–3021.
Gao B, Ahmad MF, Nagy LE, Tsukamoto H. Inflammatory pathways in alcoholic steatohepatitis. J Hepatol. 2019;70:249–59.
Zannas AS, Wiechmann T, Gassen NC, Binder EB. Gene–stress–epigenetic regulation of FKBP5: clinical and translational implications. Neuropsychopharmacology. 2015;41:261.
Garabedian MJ, Logan SK. Glucocorticoid receptor DNA binding decoy is a gas. Sci Signal. 2010;3:pe5.
Nemoto T, Kakinuma Y, Shibasaki T. Impaired miR449a-induced downregulation of Crhr1 expression in low-birth-weight rats. J Endocrinol. 2015;224:195.
Broccoli L, Uhrig S, von Jonquieres G, Schönig K, Bartsch D, Justice NJ, et al. Targeted overexpression of CRH receptor subtype 1 in central amygdala neurons: effect on alcohol-seeking behavior. Psychopharmacology. 2018;235:1821–33.
Glaser YG, Zubieta J-K, Hsu DT, Villafuerte S, Mickey BJ, Trucco EM, et al. Indirect effect of corticotropin-releasing hormone receptor 1 gene variation on negative emotionality and alcohol use via right ventrolateral prefrontal cortex. J Neurosci. 2014;34:4099.
Zorrilla EP, Heilig M, de Wit H, Shaham Y. Behavioral, biological, and chemical perspectives on targeting CRF1 receptor antagonists to treat alcoholism. Drug Alcohol Depend. 2013;128:175–86.
Hansson AC, Cippitelli A, Sommer WH, Ciccocioppo R, Heilig M. PRECLINICAL STUDY: Region-specific down-regulation of Crhr1 gene expression in alcohol-preferring msP rats following ad lib access to alcohol. Addiction Biol. 2007;12:30–34.
Treutlein J, Kissling C, Frank J, Wiemann S, Dong L, Depner M, et al. Genetic association of the human corticotropin releasing hormone receptor 1 (CRHR1) with binge drinking and alcohol intake patterns in two independent samples. Mol Psychiatry. 2006;11:594–602.
Hansson AC, Cippitelli A, Sommer WH, Fedeli A, Björk K, Soverchia L, et al. Variation at the rat Crhr1 locus and sensitivity to relapse into alcohol seeking induced by environmental stress. Proc Natl Acad Sci USA. 2006;103:15236–41.
Lucassen PJ, Pruessner J, Sousa N, Almeida OF, Van Dam AM, Rajkowska G, et al. Neuropathology of stress. Acta Neuropathol. 2014;127:109–35.
Spanagel R, Noori HR, Heilig M. Stress and alcohol interactions: animal studies and clinical significance. Trends Neurosci. 2014;37:219–27.
Clive ML, Boks MP, Vinkers CH, Osborne LM, Payne JL, Ressler KJ, et al. Discovery and replication of a peripheral tissue DNA methylation biosignature to augment a suicide prediction model. Clin Epigenetics. 2016;8:113.
Nylander I, Todkar A, Granholm L, Vrettou M, Bendre M, Boon W, et al. Evidence for a link between Fkbp5/FKBP5, early life social relations and alcohol drinking in young adult rats and humans. Mol Neurobiol. 2017;54:6225–34.
Dragan WŁ, Domozych W, Czerski PM, Dragan M. Positive metacognitions about alcohol mediate the relationship between FKBP5 variability and problematic drinking in a sample of young women. Neuropsychiatr Dis Treat. 2018;14:2681–8.
Galatzer-Levy IR, Andero R, Sawamura T, Jovanovic T, Papini S, Ressler KJ, et al. A cross species study of heterogeneity in fear extinction learning in relation to FKBP5 variation and expression: Implications for the acute treatment of posttraumatic stress disorder. Neuropharmacology. 2017;116:188–95.
Sawamura T, Klengel T, Armario A, Jovanovic T, Norrholm SD, Ressler KJ, et al. Dexamethasone treatment leads to enhanced fear extinction and dynamic Fkbp5 regulation in Amygdala. Neuropsychopharmacology. 2015;41:832.
Lee AM, Zou ME, Lim JP, Stecher J, McMahon T, Messing RO. Deletion of Prkcz increases intermittent ethanol consumption in mice. Alcohol: Clin Exp Res. 2014;38:170–8.
Wang Y, Xie Y, Li L, He Y, Zheng D, Yu P, et al. EZH2 RIP-seq identifies tissue-specific long non-coding RNAs. Curr Gene Ther. 2018;18:275–85.
Bohnsack JP, Teppen T, Kyzar EJ, Dzitoyeva S, Pandey SC. The lncRNA BDNF-AS is an epigenetic regulator in the human amygdala in early onset alcohol use disorders. Transl Psychiatry. 2019;9:34.
Acknowledgements
This research was supported by the National Institutes of Health (NIH) intramural funding ZIA-AA000242 (Section on Clinical Genomics and Experimental Therapeutics; to FWL), Division of Intramural Clinical and Biological Research of the National Institute on Alcohol Abuse and Alcoholism (NIAAA). Dr. Charlet acknowledges funding from the German Research Foundation (DFG CH1936/1-1).
The authors are grateful to the families who took part in GS, the GPs and Scottish School of Primary Care for their help in recruiting them, and the whole GS team that includes academic researchers, clinic staff, laboratory technicians, clerical workers, IT staff, statisticians and research managers. Generation Scotland received core support from the Chief Scientist Office of the Scottish Government Health Directorates (CZD/16/6) and the Scottish Funding Council (HR03006). Genotyping of the GS samples was carried out by the Genetics Core Laboratory at the Wellcome Trust Clinical Research Facility, Edinburgh, Scotland, and was funded by the Medical Research Council UK and the Wellcome Trust (Wellcome Trust Strategic Award ‘STratifying Resilience and Depression Longitudinally’ (STRADL) Reference 104036/Z/14/Z).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lohoff, F.W., Roy, A., Jung, J. et al. Epigenome-wide association study and multi-tissue replication of individuals with alcohol use disorder: evidence for abnormal glucocorticoid signaling pathway gene regulation. Mol Psychiatry 26, 2224–2237 (2021). https://doi.org/10.1038/s41380-020-0734-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-020-0734-4
This article is cited by
-
Epigenome-wide association study of alcohol use disorder in five brain regions
Neuropsychopharmacology (2022)
-
Alcohol use disorder is associated with DNA methylation-based shortening of telomere length and regulated by TESPA1: implications for aging
Molecular Psychiatry (2022)
-
Epigenome-wide association study of alcohol consumption in N = 8161 individuals and relevance to alcohol use disorder pathophysiology: identification of the cystine/glutamate transporter SLC7A11 as a top target
Molecular Psychiatry (2022)
-
Origins of human disease: the chrono-epigenetic perspective
Nature Reviews Genetics (2021)
