
Abstract
Hydrogen sulfide (H2S) is widely recognized as the third endogenous gas signaling molecule and may play a key role in cancer biological processes. ADT-OH (5-(4-hydroxyphenyl)−3H-1,2-dithiocyclopentene-3-thione) is one of the most widely used organic donors for the slow release of H2S and considered to be a potential anticancer compound. In this study, we investigated the antimetastatic effects of ADT-OH in highly metastatic melanoma cells. A tail-vein-metastasis model was established by injecting B16F10 and A375 cells into the tail veins of mice, whereas a mouse footpad-injection model was established by injecting B16F10 cells into mouse footpads. We showed that administration of ADT-OH significantly inhibited the migration and invasion of melanoma cells in the three different animal models. We further showed that ADT-OH dose-dependently inhibited the migration and invasion of B16F10, B16F1 and A375 melanoma cells as evaluated by wound healing and Transwell assays in vitro. LC-MS/MS and bioinformatics analyses revealed that ADT-OH treatment inhibited the EMT process in B16F10 and A375 cells by reducing the expression of FAK and the downstream response protein Paxillin. Overexpression of FAK reversed the inhibitory effects of ADT-OH on melanoma cell migration. Moreover, after ADT-OH treatment, melanoma cells showed abnormal expression of the H2S-producing enzymes CSE/CBS and the AKT signaling pathways. In addition, ADT-OH significantly suppressed the proliferation of melanoma cells. Collectively, these results demonstrate that ADT-OH inhibits the EMT process in melanoma cells by suppressing the CSE/CBS and FAK signaling pathways, thereby exerting its antimetastatic activity. ADT-OH may be used as an antimetastatic agent in the future.
Similar content being viewed by others

Introduction
Melanoma is the deadliest and most aggressive form of skin cancer. According to epidemiological data, 132,000 new cases of melanoma are diagnosed and 50,000 melanoma-related deaths occur worldwide each year [1]. Over the last 10 years, the rates for new cases of melanoma of the skin have been rising, on average, 1.5% each year. If melanoma is resected before metastasis, the 5-year survival rate is ~98%. However, if transfer is allowed, the survival rate is only 10%–15% [2]. Unfortunately, metastatic melanoma responds poorly to the currently available therapeutic agents. To date, there is no clinically effective drug for metastatic melanoma. Therefore, new and effective compounds are urgently needed to effectively inhibit the metastatic ability of melanoma cells and prevent their metastasis.
Hydrogen sulfide (H2S) is the third gas signaling molecule after carbon dioxide and nitric oxide that plays important roles in cancer biological processes, such as angiogenesis [3], oxidative stress [4], the cell cycle [5] and cell apoptosis [6, 7]. H2S is also endogenously produced by several enzymes, including cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), the tandem enzymes cysteine aminotransferase (CAT) and 3-mercaptopyruvate sulfurtransferase (3-MST) [8]. Given that H2S-mediated hypersulfation is important for protein function, the abnormal expression of H2S-producing enzymes can influence the development of cancer in different ways, such as tumor cell apoptosis, DNA repair, tumor growth, cancer metabolism, tumor metastasis, and angiogenesis [9, 10]. Accumulating studies have found that CSE, CBS and 3-MST play important roles in many different types of cancer cells. For example, in colon cancer, CBS, CSE and 3-MST were all observed to be significantly upregulated [3, 11, 12]. High expression levels of CSE were also found in ovarian cancer, breast cancer, thyroid cancer, and gallbladder adenocarcinoma tissues, and upregulation of CBS levels was observed in prostate cancer, gastric cancer and melanoma cells [13]. Therefore, regulating the expression of H2S-generating enzymes and changing the amount of H2S derived from the tumor could be a feasible method to change the tumor microenvironment and affect the growth and metastasis of tumor cells [14]. Although the role of endogenous H2S in tumors is still controversial, the relatively high concentration of exogenous H2S can definitely suppress the growth of cancer cells [10]. As reported, treatment with different types of H2S donors, such as DATS, GYY4137 and ATB-346, exhibits a novel anticancer effect on various kinds of tumors, including lung cancer, liver cancer, prostate cancer and other cancers [15,16,17]. This anticancer effect occurs mainly through the following mechanisms: inducing tumor cell death by increasing uncontrolled intracellular acidification [18]; inducing apoptosis by inhibiting the NF-κB pathway and STAT3 [5, 7, 15]; and triggering cell cycle arrest by inhibiting cyclin D1 [16]. It has also been reported that H2S could inhibit human breast cancer invasion and epithelial-mesenchymal transition (EMT) by reducing p38 mitogen-activated protein kinase phosphorylation [19]. However, the detailed mechanisms underlying the H2S-mediated antimetastatic effects on melanoma are still unclear, and further elucidation is needed.
We have conducted considerable research on the antitumor effects and mechanisms of H2S and its donor ADT-OH (5-(4-hydroxyphenyl)−3H-1,2-dithiocyclopentene-3-thione), which shows that the antitumor effects of the H2S donor are efficient with low toxicity. ADT-OH significantly inhibited the proliferation of a variety of tumor cells, such as A549, H446, H1299, HepG2, HCT-116 and MDA-MB-231, and especially B16F10 and A375 melanoma cells. Interestingly, ADT-OH was found to significantly inhibit melanoma cell migration, and its molecular mechanism was elucidated in this study.
Materials and methods
Plasmids, cell lines and reagents
The encoding sequence of mouse FAK was amplified from cDNA of B16F10 cells and cloned into pRK5-Flag vector. The promoter sequence of human and mouse FAK was amplified and cloned into pGL3 reporter vector just as the previous article [20]. Actinomycin D (Act D) was purchased from Sigma. B16F10 and B16F1 (murine melanoma cells), A375 (human melanoma cells) and MEF (murine embryonic fibroblast cells) were purchased from American Type Culture Collection (ATCC, USA) or maintained in our laboratory. All cells were cultured at 37 °C in 5% CO2 in a humidified atmosphere in Dulbecco’s modified Eagle’s media (DMEM, Gibco, Shanghai, China) with 10% fetal bovine serum (FBS, Gibco, Australia), penicillin (100 IU/ml) and streptomycin (100 μg/ml).
H2S measurements
B16F10 melanoma cells were seeded in 96-well plates and cultured in medium with 10 mM Cys, 10 μM PLP and ADT-OH. Lead acetate paper (RA, Sigma, St. Louis, MO, USA) was placed on the plate for 2–24 h and further incubated at 37 °C in a humidified 5% CO2 incubator. H2S detection kit (R&D Systems, Abnova, USA) was used to measure the exact amount of H2S released. B16F10 cells were treated with ADT-OH in a concentration gradient, and the cell supernatant was then collected and detected according to the manufacturer’s instructions.
Real-time quantitative PCR assay
Total RNA was extracted with TRIzol reagent (Invitrogen, USA) following the manufacturer’s instructions. Quantitative real-time PCR was performed using the reverse transcription kit (Takara, Japan) and SYBR Green PCR Master Mix (Roche, Germany). The primers were listed in Supplementary Table 1. Data were analyzed by StepOne 2.1 software (Applied Biosystems, USA) according to the manufacturer’s specifications, and were normalized using β-actin.
Protein extraction and immunoblotting
Cells were collected and homogenized with whole cell lysate prepared with RIPA lysis buffer (Santa Cruz Biotechnology) containing protease inhibitors, PMSF and orthovanadate. After 30 min of incubation on ice, the whole-cell extract was pelleted in an Eppendorf microcentrifuge and centrifuged at 12,000 r/min for 10 min at 4 °C. Supernatants were collected and protein concentration was determined by the Bio-Rad protein assay method (Bio-Rad, Hercules, CA). Proteins were separated by SDS-PAGE and transferred onto nitrocellulose membranes that were blocked with 5% non-fat milk in TBS containing 0.1% Tween-20, and incubated with primary antibodies: p-FAK (dilution 1:1000), FAK (dilution 1:1000), Akt (dilution 1:1000), p-Akt (Ser 473) (dilution 1:1000), Paxillin (dilution 1:1000), E-cadherin (dilution 1:1000), N-cadherin (dilution 1:1000), Vimentin (Cell Signaling Technology, Beverly, MA, USA; dilution 1:1000), β-actin (ABGENT, San Diego, USA; dilution 1:1000). Secondary antibodies were coupled to horseradish peroxidase, and were goat anti-rabbit or goat anti-mouse (dilution 1:2000). All experiments were done in triplicate and performed at least three times independently.
LC-MS/MS analysis and bioinformatics analysis
The LC-MS/MS analysis was performed as previously described [20]. Briefly, after collecting the cells and obtaining the lysate, the peptide was purified using a Strata-XC18 column, and then the obtained peptide was labeled with the iTRAQ Reagent-8 plex Multiplex Kit (AB Sciex U.K. Limited) according to the manufacturer’s instructions. The samples and labeled markers in this article were as follows: the samples of MEF negative control cells were labeled with iTRAQ tag 115, the samples of MEF ADT-OH (2 μM, 15 min)-treated cells were labeled with iTRAQ tag 117, the samples of MEF ADT-OH (2 μM, 2 h)-treated cells were labeled with iTRAQ tag 118, the samples of MEF ADT-OH (50 μM, 15 min)-treated cells were labeled with iTRAQ tag 119, the samples of MEF ADT-OH (50 μM, 2 h)-treated cells were labeled with iTRAQ tag 121. All the labeled samples were mixed in equal amounts and then fractionated using a Durashell C18 (5 μm, 100 Å, 4.6 × 250 mm) through high-performance liquid chromatography system (Thermo DINOEX Ultimate 3000 BioRS). Then the Triple TOF 5600 System (AB SCIEX, Concord, ON) was used for data acquisition, and ProteinPilot Software 5.0 (AB SCIEX) was used for protein identification and iTRAQ relative quantification. High abundance (>1.5-fold, P < 0.05) and low abundance (<0.67-fold, P < 0.05) proteins were used to identify differentially expressed proteins (DEPs). Finally, MetaCoreTM version 5.4 (GeneGo, St. Joseph, MI) was used to further analyze the identified DEPs through pathway analysis.
Cell cycle, cell apoptosis and cellular reactive oxygen species (ROS) levels analysis
B16F10, B16F1 and A375 cells were treated with different concentrations of ADT-OH (0, 6.3, 25, 100 μM) for 24 h. For cell cycle analysis, cells were fixed overnight at 4 °C using 70% pre-chilled ethanol (1 mL). After PI staining, cells were analyzed on a FACSCalibur flow cytometer with CellQuest software (BD Biosciences, CA, USA). For the detection of apoptosis, the treated cells were incubated with enhanced green fluorescent protein-coupled annexin V (BD Pharmingen, San Diego, CA, USA) according to the manufacturer’s instructions, and then analyzed by flow cytometry (BD Biosciences, California, USA). For the detection of ROS, the cells treated with the above drugs were resuspended in serum-free DMEM containing 10 μM MDCFH-DA and incubated at 37 °C in the dark for 30 min. After collecting and washing the cells, 400 μL of single cell suspension was analyzed using a FACSCalibur flow cytometer with CellQuest software (BD Biosciences, CA, USA). The level of intracellular reactive oxygen species was determined based on the fluorescence intensity of the FL1 channel.
Luciferase reporter assay
Cells were seeded in 12-well plates and cultured in DMEM medium with 10% FBS. After cells grew to a fusion rate of about 70%–80%, the transfection experiments were then carried out. B16F10, B16F1 and A375 cells were co-transfected with pGL3-basic, FAK-promoter luciferase (pGL3-FAK) and control pRL-SV40 reporter. Twelve hours later, vehicle and different concentrations of ADT-OH were added in the medium and the cells were cultured for another 12 h. Luciferase activities were measured consecutively by using Dual-Luciferase assays (Promega, USA). All measurements were normalized for Renilla luciferase activity to correct the variations in transfection efficiencies.
Cell proliferation assay
CCK8 assay was used to measure cell proliferation. Cells in the exponential growth phase were seeded into a 96-well plate at a density of 5000 cells per well. The cells were incubated with ADT-OH (0.8–100 μM) for 24 h before adding 10 μL of CCK8 (Sigma, Milan, Italy), and cell viability was measured using a microplate spectrophotometer (Titertek Mul-tiskan MCC/340) equipped with a 450 nm filter. Each experiment was performed in quadruplicate and repeated at least three times.
Cell migration assay
The cell migration assay was performed using transwell inserts (8.0 mm pore size, Millipore, Billerica, MA, USA). Cells were starved for 12 h prior to the experiment. Cells were then harvested and resuspended in a cell suspension diluted to 5 × 105 cells/mL with serum-free DMEM containing 1% BSA. 200 μL of cell suspension was pipetted into the upper chamber, and the lower chamber was filled with 600 μL of 10% FBS supplemented medium. After incubation at 37 °C for 10 h or 16 h, cells on the upper surface of the membrane were removed. The migrant cells attached to the lower surface were fixed in 4% paraformaldehyde at room temperature for 30 min, and stained for 30 min with a solution containing 1% crystal violet and 2% ethanol in 100 mM borate buffer (pH 9.0). The number of cells migrating to the lower surface of the membrane was photographed in five fields under a microscope with a magnification of ×100. The chamber was then purged with 33% HAC (100 μL). After the crystal violet was completely dissolved and the cells were evenly distributed in the HAC solution, the assay was performed at 570 nm using a microplate reader (TECNA, Switzerland) and quantitative analysis was performed using GraphPad Prism 8.0 software.
Wound-healing assay
Cells were plated in six-well culture plates to form cell monolayer (near 90% confluence). A sterile P-200 micropipette was used to scrape off the cells to make wounds. Then the wells were washed three times with PBS to remove non-adherent cells. The progress of wound closure was monitored with a ×10 microphotographs taken by the light microscope (Carl Zeiss Axioplan 2) at the beginning and the end of the experiments after washing with PBS. Image J software was used to calculate the scratch area of the melanoma cells treated with or without ADT-OH at 0 h and 24 h, and then the migration rates were calculated using the following formula: Migration rate = (S1 − S2)/S1, where S1 represents the scratch area at 0 h; S2 represents the scratch area at 24 h.
Colony-forming assay
Colony-forming assay was performed as previously described [21, 22]. Briefly, about 300 cells in log phase were plated into 60 mm tissue culture Petri-dish (Greiner) in triplicate with 3 mL of culture medium and cultured at 37 °C with 5% CO2. Forty-eight hours later, cells were rinsed with fresh medium, and ADT-OH was added at different concentrations (0, 3.2, 6.3, 12.5, 25, 50 μM). After incubation for 48 h, the cells were washed twice with PBS and then incubated in drug-free medium. The medium was changed every 5 days. After culturing for additional 10–14 days, the medium was discarded and each dish was washed twice with PBS carefully. The cells were fixed with methanol for 15 min and stained with a 1:10 dilution of Giemsa reagent (Merck, Germany) for 10 min. Any grouping of cells containing 30 or more cells was counted as a colony. Colony numbers were determined from triplicate plates. Colony growth was related to the control value without any treatment.
Immunofluorescence assay (IFA)
The immunofluorescence assay was performed as previously described [23]. B16F10, B16F1 and A375 cells were treated with ADT-OH at different concentrations. After 48 h, cells were harvested and immunostained with anti-E-cadherin (dilution 1:200) and Vimentin (dilution 1:200) antibodies, respectively. The cells were then incubated with Alexa Fluor 488-labeled (#A21202, Thermo Fisher Scientific Inc.; dilution 1:500) and Alexa Fluor 594-labeled (#A21207, Thermo Fisher Scientific Inc.; dilution 1:500) secondary antibody for 1 h at room temperature. After the cell nucleus was counterstained with DAPI, images were obtained using the ZEN 2009 Light Edition software (Carl Zeiss) through an inverted Zeiss LSM710 confocal microscope (40× lens; Carl Zeiss).
Development of mouse tail vein injection model
B16F10 and A375 cells (2 × 106) suspended in 200 μL of PBS were injected into the tail vein of the C57BL/6 mice or nude mice. Beginning on the second day, the mice were administered with vehicle, 17.5 mg/kg ADT-OH, 37.5 mg/kg ADT-OH, 75 mg/kg ADT-OH, 1.4 mg/kg NaHS, 2.8 mg/kg NaHS or 5.6 mg/kg NaHS five times a week for a total of 3 weeks with at least eight mice per group. The mice were then repeatedly imaged for metastatic tumor spreading to distant organs. At the end of the study, mice were euthanized and the lungs and livers were harvested, fixed in 10% formalin, and paraffin embedded for pathological examination of H&E slides.
In vivo footpad-injection model
B16F10 cells (2 × 105 cells in 50 μL PBS) were implanted into the right hind footpads of C57BL/6 mice using a Hamilton syringe and 25‐gauge needle. The detailed treatment was described in Fig. 1b. All mice were euthanized 21 days after tumor inoculation in order to assess the number of spontaneous metastases in the lungs. The following formula was used to measure and calculate the volume of the tumor in the sole of the foot: 0.5236 × L1 × (L2)2, where L1 is the long axis of the tumor and L2 is the short axis of the tumor.

a The levels of the CBS and CSE genes are associated with the prognosis of patients with different cancers (bladder urothelial carcinoma, lung adenocarcinoma and skin cutaneous melanoma). Kaplan–Meier curves for recurrence-free survival were created using a Kaplan–Meier plotter (www.kmplot.com), in which bladder urothelial carcinoma, lung adenocarcinoma and skin cutaneous melanoma patients were classified according to high and low CBS/CSE gene expression. The hazard ratio (with 95% confidence interval) and the log rank P value were calculated. b Animal treatment methods. c Representative imaging of footpad model mice and the tumor volume at the footpad of each group at the end of the experiment (n = 6). Data are represented as mean ± SD. *P < 0.05, **P < 0.01. d Representative H&E tissue staining of lungs from footpad model mice.
Hematoxylin-Eosin staining assay
In a parallel animal assay (totally three groups, and three mice per group), the tumor establishment and drug treatment are the same as described above. On the 21th day, mice were euthanized. Tumors were collected, fixed with 4% formaldehyde and then embedded in paraffin. Tissue sections (5 μm in thickness) were prepared according to standard protocols for hematoxylin/eosin (H&E) staining.
Statistical analysis
Data were presented as means ± SD. Comparisons within groups were done with a t-test with repeated measures; P values indicated in Figures are <0.05 (*), <0.01 (**), <0.005 (***), and <0.001 (****).
Results
ADT-OH inhibits melanoma invasion in vivo
H2S is produced in mammalian cells by three major enzymes, CSE, CBS, and 3-MST. Numerous studies have shown that the expression of these enzymes changes in various types of cancer cell processes, especially CSE and CBS [3, 24,25,26]. According to Oncomine, an online cancer transcriptome database, CSE and CBS were overexpressed in human skin cutaneous melanoma (SKCM). Kaplan–Meier survival analysis of SKCM patients was performed based on the patient data in TCGA. In addition, the results showed that patients with higher expression of CSE or CBS exhibited poorer overall survival (Fig. 1a). Furthermore, we found that the overall survival rates of patients exhibiting high CSE and CBS expression with lung adenocarcinoma, acute myeloid leukemia, bladder urothelial carcinoma, and adrenocortical carcinoma were also significantly reduced based on The Cancer Genome Atlas (TCGA) datasets (Fig. 1a, Supplementary Fig. 1 and Supplementary Fig. 2). In our study, after treatment with ADT-OH, the protein levels of CBS and CSE in melanoma cells were found to be significantly downregulated (Supplementary Fig. 3). Through a specific reaction, H2S and lead acetate form a black precipitate (lead sulfide), which can be captured on filter paper containing lead acetate. As shown in Supplementary Fig. 4, we found that ADT-OH released H2S in a dose-dependent manner, indicating that ADT-OH is a slow-releasing organic H2S donor. Moreover, Supplementary Fig. 5 shows that ADT-OH did not significantly induce apoptosis in melanoma cells at low concentrations (<12.5 μM); however, when the concentration exceeded 12.5 μM, ADT-OH treatment significantly induced the apoptosis of melanoma cells. In addition, real-time cell analysis was also employed to detect the dynamic proliferation kinetic curve of melanoma cells. As shown in Supplementary Fig. 6, the results also showed that ADT-OH treatment at low concentrations had no apparent side effect on the proliferation rate of melanoma cells. Taken together, these results reveal that H2S plays a vital role in the molecular biological characteristics of cancer and may be a potential drug target to improve prognosis. Therefore, a palm transfer model was implemented. As shown in Fig. 1b and c, intraplantar injection of mouse melanoma cells (5 × 105 cells in 20 μL) into C57BL/6 mice led to time-dependent tumor growth in the hind paw, showing a 3–4-fold increase in paw volume 3 weeks after melanoma cell implantation. After treatment with ADT-OH, the paw volume of melanoma-bearing mice decreased from 252.128 mm3 to 105.2557 mm3. Moreover, H&E staining analysis showed that ADT-OH treatment significantly reduced the number of melanoma nodules in the lungs of mice, especially after treatment with a dose of 37.5 mg/kg (Fig. 1d). Furthermore, a mouse passive lung metastasis model was established to investigate the effects of ADT-OH on melanoma cancer metastasis in vivo (Fig. 2a). Lung metastasis nodules were found in the lungs and counted macroscopically (Fig. 2b). Compared with the control group, lung metastasis was restrained in the ADT-OH- and NaHS-treated groups, but especially in the ADT-OH (18.75 and 37.5 mg/kg)-treated groups. Likewise, the H&E staining results in Fig. 2c show that ADT-OH and NaHS treatment could significantly decrease B16F10 cell lung metastases and reduce lung nodule volume. We also performed the same experiment on female BALB/c nude mice using A375 cells. As shown in Fig. 2d and e, A375 cell lung metastases and lung nodule volumes were markedly decreased after ADT-OH treatment. Taken together, our study suggests that ADT-OH could significantly inhibit the migration and invasion of melanoma cells in vivo.

a Animal treatment methods. b Representative lung imaging of mice after tail vein injection with B16F10 cells for 3 weeks. c H&E stained lung sections. The representative images showed lung tumor distribution of the lung metastasis model mice treated with vehicle, 18.75 mg/kg ADT-OH, 37.5 mg/kg ADT-OH, 75 mg/kg ADT-OH, 1.4 mg/kg NaHS, 2.8 mg/kg NaHS or 5.6 mg/kg NaHS. Scale bars, 100 μm. d–e Representative lung imaging (d) and H&E tissue staining (e) of mice injected with A375 cells via the tail vein. The lung metastasis model mice were treated with vehicle, 37.5 mg/kg ADT-OH, 75 mg/kg ADT-OH or 2.8 mg/kg NaHS for 3 weeks. Scale bars, 100 μm.
ADT-OH suppresses melanoma cell migration and invasion in vitro
Migration plays a vital role in the process of tumor cell metastasis [27]. Due to the lack of effective therapies for metastatic melanoma in clinical practice and the antimetastatic activity exhibited by ADT-OH in vivo, we next evaluated the antimigratory effects of ADT-OH on A375, B16F10 and B16F1 metastatic melanoma cells in vitro. As shown in Fig. 3a, d, the wound healing assay showed that 6.3 μM ADT-OH significantly reduced the migration of B16F10 cells, and ADT-OH also showed the same effects on B16F1 and A375 melanoma cells (Fig. 3b, e, c, f). However, in the absence of ADT-OH, melanoma cells (including A375, B16F10 and B16F1 cells) displayed high migratory capabilities, which was indicated by their ability to completely heal the scratch wound (Fig. 3). Taken together, these results indicated that ADT-OH significantly suppressed the migratory activities of A375, B16F10 and B16F1 melanoma cells in a dose-dependent manner.

a–c Melanoma cell lines B16F10, B16F1 and A375 were seeded into six-well plates at 2 × 105 cells/well. After incubation with ADT-OH at 0 μM (control), 6.3 μM, 12.5 μM, 50 μM for 24 h, the effect of ADT-OH on cell migration was measured by wound-healing assay; original magnification ×40. d–f The migration rates of B16F10, B16F1 and A375 cells were calculated by the formula shown in Materials and Methods. Data are presented as mean ± SD of three independent experiments; *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001 compared with vehicle group.
ADT-OH exhibits anti-invasive activity in melanoma cells in vitro
Tumor metastasis is the final biological process of the invasion-metastasis cascade [28], and tumor cell invasion also plays crucial roles during tumor cell metastasis. Matrigel-coated Transwell chamber assays are usually used to examine cell invasion ability. To explore whether ADT-OH can inhibit the invasion ability of A375, B16F10 and B16F1 cells, we performed an invasion assay following ADT-OH treatment. The results showed that in the absence of ADT-OH, A375, B16F10 and B16F1 cells completely penetrated the filter membrane coated with Matrigel, showing a high degree of invasion. However, the invasion capabilities of A375, B16F10 and B16F1 cells were effectively suppressed by ADT-OH treatment in a dose-dependent manner (Fig. 4a–c). To further visualize the effects of ADT-OH on the actin cytoskeletons of melanoma cells, a phalloidin immunofluorescence staining assay was performed. We observed that ADT-OH treatment clearly inhibited filopodia production and spear elongation of A375, B16F10 and B16F1 cells (Fig. 4d–f). Taken together, these data indicated that ADT-OH could reduce the migration and invasion capability of melanoma cells in vitro.

a–c Summary of results from the transwell invasion assay. After 12 h of incubation with different concentrations of ADT-OH (0, 6.3, 25, 100 μM), the cells that migrated to the lower chamber were fixed, stained, and counted using a light microscope. Four random fields per filter were scanned for the presence of cells on the lower side of the membrane. The panels of the upper figures show the images obtained by cell invasion assay in B16F10, B16F1 and A375 cells. All experiments were performed thrice in triplicate. The data are presented as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.005 compared with vehicle group. d–f Representative images of F-actin immunofluorescence in B16F10, B16F1 and A375 cells. The cells were treated under the same experimental conditions as (a–c). Scale bars correspond to 20 μm.
ADT-OH inhibits cell migration by regulating the FAK signaling pathway
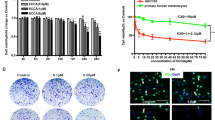
To further explore the specific role of ADT-OH on cell migration and the signaling pathway that might be involved, we performed high-throughput proteomic screen to compare protein expression levels between ADT-OH-treated and ADT-OH-untreated cell lines. In total, 2217 DEPs in all groups were screened by a t-test (Supplementary file 3). The MetaCoreTM pathway mapping tool clustered the differential networks from the DEP results, among which the cell adhesion and migration pathways showed a high score (Fig. 5a, b). Moreover, as shown in Supplementary Fig. 7, the intracellular protein changes under the action of 2 μM and 50 μM ADT-OH were quite different, of which only 165 proteins had the same change patterns. Next, further analysis of the process networks of the two groups found that the 2 μM ADT-OH group was more closely related to tumor metastasis. As shown in Supplementary Fig. 7c, the fourth cell pathway after 2 μM ADT-OH treatment was cell migration and adhesion. The FAK signaling pathway plays a crucial role in the metastasis and invasion of a variety of tumor cells [29,30,31]. Here, we found that FAK was involved in multiple signaling pathways, including cell migration, following ADT-OH treatment (Fig. 5c and Supplementary Fig. 8). To better determine the cell migration or adhesion-related signaling pathways, we reanalyzed the potentially changed pathway maps in the 2 μM ADT-OH-treatment group with MetaCoreTM GeneGo Pathway Maps software. In Supplementary Fig. 9, FAK was shown to be the key changed protein involved in the cell adhesion-related signaling pathways. In addition, as shown in Fig. 5d and Supplementary Fig. 10, ADT-OH might directly affect cell migration through the PTEN/FAK/Paxillin pathway. Therefore, the changes in protein and mRNA levels in this signaling pathway after ADT-OH treatment were next detected by Western blotting and qPCR analyses. As shown in Fig. 6a and b, both total FAK expression and its phosphorylation levels decreased greatly in melanoma cells in response to ADT-OH treatment. After ADT-OH treatment, the protein level of Paxillin, the downstream response protein of FAK, was also significantly reduced. In addition, the occurrence of EMT programs is often accompanied by the loss of cell epithelial characteristics and the acquisition of mesenchymal characteristics, showing enhanced motility and invasiveness. Next, a Western blot assay was used to detect the regulatory effects of ADT-OH on the protein expression levels of E-cadherin, N-cadherin and Vimentin, all of which are typical markers of the EMT process. We found that ADT-OH significantly reinduced the protein expression level of E-cadherin and reinhibited the protein expression levels of N-cadherin and Vimentin compared to the vehicle group (Fig. 6a, b, Supplementary Fig. 11 and Supplementary Fig. 12). The above results indicated that ADT-OH significantly reversed the EMT process in A375 and B16F10 cells and thus inhibited tumor cell metastasis. Furthermore, the change patterns in the mRNA levels of FAK, Paxillin, E-cadherin and N-cadherin were similar to those of their protein levels (Supplementary Fig. 13). Immunofluorescence assays were also performed to show that ADT-OH-treated melanoma cells exhibited suppressed epithelial markers, such as E-cadherin, and overexpressed mesenchymal markers, such as Vimentin, in comparison with the control (Fig. 6c, d, Supplementary Fig. 14 and Supplementary Fig. 15).

a MetaCoreTM GeneGo Pathway Maps was used to analyze the ontological categories of differentially expressed proteins in MEF cells treated with or without ADT-OH. b The protein network associated with cell adhesion and migration is differentially expressed in MEF cells treated with ADT-OH. Higher definition image is shown in Supplementary Fig. 8. c The network objects involved in Pathway Maps analyzed by MetaCoreTM GeneGo Pathway Maps software. d The GeneGO pathway shows changes in the expression of proteins involved in FAK/Paxillin signaling. The various proteins on the map are represented by different symbols (representing the functional categories of proteins). Higher definition image is shown in Supplementary Fig. 10. The symbol next to the thermometer shaded in blue or red indicates the protein identified in this study: blue indicates the protein that is downregulated in ADT-OH treated MEF cells relative to MEF negative control cells (treated with vehicle). Red represents up-regulated protein.

a–b Western blot analysis of p-FAK, FAK, Paxillin, p-AKT, AKT, E-Cadherin, N-Cadherin and Vimentin in B16F10 (a) and A375 (b) cells treated with ADT-OH (24 h) at different concentrations. c–d Representative immunofluorescence staining for Vimentin (red) and E-Cadherin (green) in B16F10 (c) and A375 (d) cells treated with different concentrations of ADT-OH for 24 h. The nuclei were stained with DAPI. Clear Figures are shown in Supplementary Fig. 14. Scale bars correspond to 50 μm.
ADT-OH affects the protein level of FAK mainly by decreasing its stability
As shown in Fig. 7a, ADT-OH significantly reduced the FAK mRNA levels in B16F10, B16F1 and A375 cells. To explore the mechanism of ADT-OH on FAK, we constructed a FAK luciferase reporter gene plasmid and performed experiments in B16F10, B16F1 and A375 melanoma cells. Detection of the dual luciferase reporter gene showed that ADT-OH had no obvious effect on the transcriptional activity of the FAK promoter (Fig. 7b). However, mRNA stability experiments showed that ADT-OH significantly reduced the stability of FAK mRNA (Fig. 7c). Therefore, we speculate that ADT-OH might inhibit cell migration by reducing the level of FAK mRNA and subsequently reducing its protein expression level.

a The FAK mRNA levels of B16F10, B16F1 and A375 cells were detected by qPCR analysis after ADT-OH treatment for 6 h, respectively. b Detection of FAK promoter transcriptional activity after ADT-OH treatment with dual luciferase in B16F10, B16F1 and A375 cells. c B16F10, B16F1 and A375 cells were treated with or without Act D (0.05 μg/mL) and ADT-OH (6.3 μM) for 12 h, and then the mRNA levels of FAK were detected. *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001. Data are expressed as mean ± SD of three independent experiments.
Then, we determined whether FAK overexpression could reverse the inhibitory effects of ADT-OH on melanoma migration. B16F10 cells were transfected with the FAK-overexpression vector (Fig. 8). We found that the FAK mRNA and protein expression levels increased significantly in the transfected cells (Fig. 8a, b). Compared with negatively transfected cells, overexpression of FAK significantly promoted the protein and mRNA expression of N-cadherin and Vimentin. In contrast to the cells that were not treated with either ADT-OH or NaHS, the expression levels of E-cadherin significantly increased and the expression levels of FAK, p-AKT, and AKT significantly decreased in B16F10 cells after ADT-OH or NaHS treatment, along with the microvilli and pseudopodia; however, these effects and this phenotype were significantly abolished after FAK overexpression (Fig. 8a, b). As shown in Fig. 8c, the level of F-actin in melanoma cells decreased, the cell structure was disorganized, and the cell morphology became round after treating control cells with ADT-OH. However, cells overexpressing FAK showed increased levels of the F-actin protein, the cells became elongated and grew fusiform, and the barbed ends of the cells were more prominent. After NaHS or ADT-OH treatment, this melanoma cell phenotype did not show a clear change. These results indicate that by reducing FAK expression, ADT-OH significantly inhibited the EMT process of melanoma, whereas this ADT-OH-induced effect was significantly reversed after FAK overexpression. The migratory potency of cells is inhibited once the EMT process is suppressed in melanoma cells. The results of the Transwell experiment further showed that the migration ability of B16F10 cells was significantly inhibited after ADT-OH treatment but restored after FAK overexpression (Fig. 8d).

a B16F10 cells were transfected with pRK5-FAK or pRK5 plasmid for 24 h and then treated with NaHS (2 mM) or ADT-OH (6.3 μM) for 24 h. The cell lysates were performed by Western blot with indicated antibodies. b The mRNA levels of FAK, E-cadherin, N-cadherin and Vimentin in B16F10 cells under different treatment conditions were examined by RT-qPCR analysis. c After transfected with pRK5-FAK or pRK5 plasmid, B16F10 cells were treated with NaHS (2 mM) or ADT-OH (6.3 μM) for 24 h. Immunofluorescence staining for FAK and F-actin was performed and representative images are shown. DAPI was used to display the nucleus of cells. d Transwell assay. B16F10 cells were transfected with pRK5-FAK or pRK5 plasmid and then exposed to NaHS (2 mM) or ADT-OH (6.3 μM). After 24 h, the cells that migrated to the inferior membrane were stained and counted in five fields at a magnification of ×100. n = 5, bar = 100 μm. The experiments were carried out in triplicate and representative data are shown. *P < 0.05, **P < 0.01, ***P < 0.005
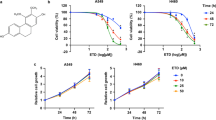
ADT-OH inhibits the viability of melanoma cells in vitro
To investigate the cytotoxicity of ADT-OH, we next applied a CCK-8 assay to evaluate the inhibitory effects of ADT-OH on the proliferation of B16F10 and A375 cells and MEFs. Supplementary Fig. 16 shows that ADT-OH inhibited the viability of A375 cells and B16F10 cells in both a time- and concentration-dependent manner but had only a slight effect on MEFs. After pretreatment with ADT-OH for 24 h, ADT-OH exhibited an IC50 value of 11.67 μM against A375 cells and an IC50 value of 5.653 μM against B16F10 cells, while the IC50 value against MEFs was 32.37 μM. Furthermore, we found that there were no obvious cytotoxic effects on A375 cells, B16F10 cells or MEFs when these cells were treated with 0–6.4 μM ADT-OH (Supplementary Fig. 16). In addition, the CCK-8 assay showed that ADT-OH could significantly inhibit the proliferation of a variety of tumor cells other than melanoma (Supplementary Fig. 17). Next, the long-term viability of ADT-OH-treated melanoma cells and control cells was tested with a colony formation assay (Supplementary Fig. 18). The results showed that ADT-OH significantly inhibited the colony formation capacity of melanoma cells. Moreover, we studied whether ADT-OH had any effects on the cell cycle in B16F10, B16F1 and A375 cells. As shown in Supplementary Fig. 19, ADT-OH arrested these tumor cells in the G2/M phase, while the effect of NaHS treatment on the cell cycle was not significant. Analysis of apoptosis further confirmed that ADT-OH at relatively higher concentrations promoted apoptosis of B16F10, B16F1 and A375 cells (Supplementary Fig. 5 and Supplementary Fig. 19). In addition, ADT-OH increased the contents of ROS in melanoma cells to further promote tumor cell death (Supplementary Fig. 20). These results indicate that higher concentrations of ADT-OH can not only inhibit the migration of melanoma cells but also inhibit cell activity by hindering the melanoma cycle and proliferation, thereby achieving a more effective melanoma treatment.
Discussion
Melanoma is a highly aggressive cancer, and tumor metastasis is the main cause of patient death from this disease. At the time of diagnosis, many cancer patients have already experienced metastasis, leading to high lethality [32]. Tumor metastasis is a complicated biological process involving multiple successive and interrelated steps, such as cell separation, adhesion, invasion, and migration [33]. Unfortunately, there is still a lack of effective drugs to treat melanoma metastasis in clinical practice. Currently, various H2S donors, including ADT-OH, have been confirmed to inhibit the proliferation and development of melanoma [15, 23]. An increasing number of research studies have focused on elucidating the molecular mechanisms of the anticancer effects of H2S, and several critical signaling pathways have been reported to be influenced by treatment with H2S donors [34, 35]. The multiple functional effects of H2S administration in cancer cells involve the induction of apoptosis, the inhibition of proliferation and the prevention of metastasis and tumor angiogenesis [15, 16, 36,37,38]. H2S is mainly synthesized in the cell through the trans-sulfurase reaction involving CBS and CSE. Endogenous H2S in mitochondria is produced by 3-MST in the presence of 3-mercaptopyruvate. CBS catalyzes H2S by driving beta replacement and is a constitutively expressed enzyme whose activity can be adjusted after translation. Current research reported that increased CBS levels have been observed in breast cancer, ovarian cancer, colon cancer, thyroid cancer, gallbladder adenocarcinoma tissues and others [13, 39]. CSE is another H2S-producing enzyme that shows significantly different expression levels in several different cancer types. For example, in prostate cancer, the overexpression of CSE increases the production of endogenous H2S and leads to nuclear factor-κB (NF-κB)-mediated activation of interleukin 1β signals, thereby resulting in the enhancement of cell invasion, angiogenesis, lymphangiogenesis, tumor growth and cell metastasis in prostate cancer [40]. In addition, it has been reported that by regulating the expression of H2S-producing enzymes and treatment with H2S donors, the amount of tumor-derived H2S will be changed, thereby exhibiting anticancer effects [10]. Therefore, an in-depth understanding of the mechanism by which H2S donors inhibit the metastatic ability of melanoma will provide new therapeutics for melanoma metastasis. In addition, the slow-releasing H2S donor 5-(4-hydroxyphenyl)−3H-1,2-dithiocyclopentene-3-thione (ADT-OH) is known as a potential therapeutic with chemopreventive and cytoprotective properties due to its 3H-1,2-dithiole-3-thione group (dithiolethione), which is also one of the most widely used moieties for synthesizing slow-releasing organic H2S donors [36, 41]. Previously, we found that ADT-OH is a potential antitumor agent that enhances the FADD-dependent extrinsic apoptosis of melanoma cells [23]. Here, we further broaden the potential antitumor effects of ADT-OH by demonstrating its ability to inhibit melanoma metastasis both in vitro and in vivo.
A375 is a human melanoma cell line, whereas B16F10 and B16F1 are mouse melanoma cell lines. In the current study, these cell lines were selected to explore the antimetastatic effects of ADT-OH. We first found that ADT-OH significantly inhibited melanoma cell migration in vivo in a palm transfer model and a mouse passive lung metastasis model. Similarly, ADT-OH significantly suppressed the migration and invasion of lung cancer cells in vitro. Further research found that ADT-OH inhibited the proliferation of A375, B16F10 and B16F1 melanoma cells but exhibited little toxicity to normal MEFs. Moreover, the CCK-8 assay and flow cytometry assay were used to detect the viability of ADT-OH-treated melanoma cells at different concentrations. Interestingly, our results showed that the main effect of ADT-OH at relatively low concentrations was suppression of the migration ability of melanoma cells with few inhibitory effects on cell proliferation; however, when the concentration exceeded 12.5 μM, the main effect of ADT-OH was the inhibition of melanoma cell proliferation. In addition, proteomics data showed that a low concentration of ADT-OH is more closely related to tumor migration, while a high concentration of ADT-OH shows a greater effect on cell apoptosis and survival. In addition, in vitro experiments further showed that ADT-OH at a low concentration (such as 6.3 μM) can significantly inhibit the metastasis of melanoma cells but cannot cause clear melanoma cell apoptosis. Collectively, these results indicated that the inhibition of tumor cell migration caused by ADT-OH at relatively low concentrations is not achieved by promoting tumor cell apoptosis.
The EMT process plays a critical role in regulating tumor metastasis. This cellular phenomenon is characterized by the loss of typical epithelial characteristics, including cell polarity, intercellular adhesion and the acquisition of mesenchymal properties [42]. Intriguingly, the EMT process is involved in various tumor functions, including tumor metastasis, tumor cell migration, tumor initiation, tumor stemness, malignant progression, and therapeutic resistance [43]. Many studies have confirmed that EMT plays a decisive role in tumor progression and metastasis [42, 44,45,46]. In this study, we observed that ADT-OH increased the expression levels of epithelial markers (such as E-cadherin) and decreased the expression levels of mesenchymal markers (such as N-cadherin and Vimentin) in melanoma cells. These findings suggest that ADT-OH negatively regulates the EMT process in melanoma cells and thereby significantly weakens tumor cell migration, invasion, and adhesion in vitro. Currently, many investigations have revealed that H2S inhibits the EMT process induced by TGF-β1 in A549 and HK-2 cells [38, 47]. Therefore, the antimetastatic effects of ADT-OH on melanoma cells mediated by inhibition of the EMT process in this study are consistent with these previously reported works.
FA kinase (FAK) is a nonreceptor tyrosine kinase that participates in FA complex formation. It has been found to be dysregulated in various types of cancer in relation to tumor metastasis [48,49,50,51]. Paxillin, which is a structural protein of the FA complex, also contributes to metastasis [52]. FAK, a key regulator of integrin-mediated adhesion, can be autophosphorylated at Tyr-397 upon recruitment of this kinase to FA sites following the binding of the transmembrane integrin receptor to the ECM. Activated FAK provides both signal transduction and scaffolding functions. Paxillin is an important FA-associated cytoskeletal adaptor protein that provides a docking site for FAK. In turn, FAK can phosphorylate Paxillin at Tyr-118 to regulate its functions. Phosphorylation of both proteins is required for FA formation, cell motility, and metastasis. Therefore, targeted inhibition of FAK signaling pathways has been considered a promising strategy for melanoma metastasis therapy. Previous studies have demonstrated that H2S and H2S donors covalently react with functional proteins to affect a variety of signaling pathways, including the NF-κB, EGF, PI3K/Akt, MAPK and integrin pathways [47, 53, 54]. However, the role of ADT-OH in melanoma cell adhesion and the involvement of FAK or Paxillin in this biological process remain largely unknown. In this study, ADT-OH treatment decreased the expression levels and phosphorylation levels of FAK in a dose-dependent manner in both A375 and B16F10 cells. In contrast, overexpression of FAK reversed these effects and restored the phenotype exerted by ADT-OH in melanoma cells, suggesting the critical role of the FAK signaling pathway in the ADT-OH-induced inhibitory effects on cell migration.
Furthermore, ADT-OH decreased the expression of CSE and CBS, which further elucidated the mechanism by which ADT-OH inhibits melanoma migration. Therefore, we speculated that the inhibitory effects of ADT-OH on melanoma metastasis may be partly attributed to the ability of ADT-OH to inhibit the CSE/CBS and FAK/Paxillin signaling pathways. To the best of our knowledge, this is the first demonstration of the antimetastatic mechanism of ADT-OH in melanoma cells.
In summary, our results indicate that ADT-OH achieves its potent antitumor metastasis effects by suppressing the CSE/CBS and FAK/Paxillin signaling pathways in vivo and in vitro, thereby inhibiting the EMT process (Supplementary Fig. 21). Moreover, ADT-OH shows reduced cytotoxicity to normal cell lines (such as MEFs and 2B cells). Together, our findings may provide new potential therapies for inhibiting melanoma metastasis. However, more in-depth research is needed to determine the detailed mechanism and potential of ADT-OH with respect to its antitumor effects, and it remains to be explored whether the H2S released by ADT-OH or ADT-OH itself causes its improved antitumor activity. In our future research, high-throughput screening analysis and other methods will be employed to explore the potential targets on which ADT-OH exerts its antitumor effects.
References
Geller AC, Clapp RW, Sober AJ, Gonsalves L, Mueller L, Christiansen CL, et al. Melanoma epidemic: an analysis of six decades of data from the Connecticut Tumor Registry. J Clin Oncol. 2013;31:4172–8.
Palrasu M, Knapinska AM, Diez J, Smith L, LaVoi T, Giulianotti M, et al. A novel probe for spliceosomal proteins that induces autophagy and death of melanoma cells reveals new targets for melanoma drug discovery. Cell Physiol Biochem. 2019;53:656–86.
Szabo C, Coletta C, Chao C, Modis K, Szczesny B, Papapetropoulos A, et al. Tumor-derived hydrogen sulfide, produced by cystathionine-beta-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc Natl Acad Sci USA. 2013;110:12474–9.
Arribillaga L, Dotor J, Basagoiti M, Riezu-Boj JI, Borras-Cuesta F, Lasarte JJ, et al. Therapeutic effect of a peptide inhibitor of TGF-beta on pulmonary fibrosis. Cytokine. 2011;53:327–33.
Lu S, Gao Y, Huang X, Wang X. GYY4137, a hydrogen sulfide (H2S) donor, shows potent anti-hepatocellular carcinoma activity through blocking the STAT3 pathway. Int J Oncol. 2014;44:1259–67.
Murata T, Sato T, Kamoda T, Moriyama H, Kumazawa Y, Hanada N. Differential susceptibility to hydrogen sulfide-induced apoptosis between PHLDA1-overexpressing oral cancer cell lines and oral keratinocytes: role of PHLDA1 as an apoptosis suppressor. Exp Cell Res. 2014;320:247–57.
Panza E, De Cicco P, Armogida C, Scognamiglio G, Gigantino V, Botti G, et al. Role of the cystathionine gamma lyase/hydrogen sulfide pathway in human melanoma progression. Pigment Cell Melanoma Res. 2015;28:61–72.
Kashfi K, Olson KR. Biology and therapeutic potential of hydrogen sulfide and hydrogen sulfide-releasing chimeras. Biochem Pharmacol. 2013;85:689–703.
Ngowi EE, Afzal A, Sarfraz M, Khattak S, Zaman SU, Khan NH, et al. Role of hydrogen sulfide donors in cancer development and progression. Int J Biol Sci. 2021;17:73–88.
Wu D, Si W, Wang M, Lv S, Ji A, Li Y. Hydrogen sulfide in cancer: Friend or foe? Nitric Oxide. 2015;50:38–45.
Fan K, Li N, Qi J, Yin P, Zhao C, Wang L, et al. Wnt/beta-catenin signaling induces the transcription of cystathionine-gamma-lyase, a stimulator of tumor in colon cancer. Cell Signal. 2014;26:2801–8.
Olah G, Modis K, Toro G, Hellmich MR, Szczesny B, Szabo C. Role of endogenous and exogenous nitric oxide, carbon monoxide and hydrogen sulfide in HCT116 colon cancer cell proliferation. Biochem Pharmacol. 2018;149:186–204.
Wang RH, Chu YH, Lin KT. The hidden role of hydrogen sulfide metabolism in cancer. Int J Mol Sci. 2021;22:6562.
Giuffre A, Tome CS, Fernandes DGF, Zuhra K, Vicente JB. Hydrogen sulfide metabolism and signaling in the tumor microenvironment. Adv Exp Med Biol. 2020;1219:335–53.
De Cicco P, Panza E, Ercolano G, Armogida C, Sessa G, Pirozzi G, et al. ATB-346, a novel hydrogen sulfide-releasing anti-inflammatory drug, induces apoptosis of human melanoma cells and inhibits melanoma development in vivo. Pharmacol Res. 2016;114:67–73.
Lee ZW, Zhou J, Chen CS, Zhao Y, Tan CH, Li L, et al. The slow-releasing hydrogen sulfide donor, GYY4137, exhibits novel anti-cancer effects in vitro and in vivo. PLoS One. 2011;6:e21077.
Yi L, Su Q. Molecular mechanisms for the anti-cancer effects of diallyl disulfide. Food Chem Toxicol. 2013;57:362–70.
Lee ZW, Teo XY, Tay EY, Tan CH, Hagen T, Moore PK, et al. Utilizing hydrogen sulfide as a novel anti-cancer agent by targeting cancer glycolysis and pH imbalance. Br J Pharmacol. 2014;171:4322–36.
Lv M, Li Y, Ji MH, Zhuang M, Tang JH. Inhibition of invasion and epithelial-mesenchymal transition of human breast cancer cells by hydrogen sulfide through decreased phospho-p38 expression. Mol Med Rep. 2014;10:341–6.
Jiang W, Cai F, Xu H, Lu Y, Chen J, Liu J, et al. Extracellular signal regulated kinase 5 promotes cell migration, invasion and lung metastasis in a FAK-dependent manner. Protein Cell. 2020;11:825–45.
Cai F, Chen M, Zha D, Zhang P, Zhang X, Cao N, et al. Curcumol potentiates celecoxib-induced growth inhibition and apoptosis in human non-small cell lung cancer. Oncotarget. 2017;8:115526–45.
Jiang W, Jin G, Cai F, Chen X, Cao N, Zhang X, et al. Extracellular signal-regulated kinase 5 increases radioresistance of lung cancer cells by enhancing the DNA damage response. Exp Mol Med. 2019;51:1–20.
Cai F, Xu H, Cao N, Zhang X, Liu J, Lu Y, et al. ADT-OH, a hydrogen sulfide-releasing donor, induces apoptosis and inhibits the development of melanoma in vivo by upregulating FADD. Cell Death Dis. 2020;11:33.
Guo H, Gai JW, Wang Y, Jin HF, Du JB, Jin J. Characterization of hydrogen sulfide and its synthases, cystathionine beta-synthase and cystathionine gamma-lyase, in human prostatic tissue and cells. Urology. 2012;79:483 e1–5.
Zhang L, Qi Q, Yang J, Sun D, Li C, Xue Y, et al. An anticancer role of hydrogen sulfide in human gastric cancer cells. Oxid Med Cell Longev. 2015;2015:636410.
Jurkowska H, Placha W, Nagahara N, Wrobel M. The expression and activity of cystathionine-gamma-lyase and 3-mercaptopyruvate sulfurtransferase in human neoplastic cell lines. Amino Acids. 2011;41:151–8.
Heerboth S, Housman G, Leary M, Longacre M, Byler S, Lapinska K, et al. EMT and tumor metastasis. Clin Transl Med. 2015;4:6.
Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275–92.
Cheng Y, Gao XH, Li XJ, Cao QH, Zhao DD, Zhou JR, et al. Depression promotes prostate cancer invasion and metastasis via a sympathetic-cAMP-FAK signaling pathway. Oncogene. 2018;37:2953–66.
Li R, Wang Y, Zhang X, Feng M, Ma J, Li J, et al. Exosome-mediated secretion of LOXL4 promotes hepatocellular carcinoma cell invasion and metastasis. Mol Cancer. 2019;18:18.
Jiang WG, Sanders AJ, Katoh M, Ungefroren H, Gieseler F, Prince M, et al. Tissue invasion and metastasis: Molecular, biological and clinical perspectives. Semin Cancer Biol. 2015;35:S244–S75. Suppl
Mishra H, Mishra PK, Ekielski A, Jaggi M, Iqbal Z, Talegaonkar S. Melanoma treatment: from conventional to nanotechnology. J Cancer Res Clin Oncol. 2018;144:2283–302.
Pal M. Tumor metastasis suppressor functions of Ets transcription factor through integrin beta3-mediated signaling pathway. J Cell Physiol. 2019;234:20266–74.
Chen S, Yue T, Huang Z, Zhu J, Bu D, Wang X, et al. Inhibition of hydrogen sulfide synthesis reverses acquired resistance to 5-FU through miR-215-5p-EREG/TYMS axis in colon cancer cells. Cancer Lett. 2019;466:49–60.
Sakuma S, Minamino S, Takase M, Ishiyama Y, Hosokura H, Kohda T, et al. Hydrogen sulfide donor GYY4137 suppresses proliferation of human colorectal cancer Caco-2 cells by inducing both cell cycle arrest and cell death. Heliyon. 2019;5:e02244.
Yang CT, Chen L, Xu S, Day JJ, Li X, Xian M. Recent development of hydrogen sulfide releasing/stimulating reagents and their potential applications in cancer and glycometabolic disorders. Front Pharmacol. 2017;8:664.
Chattopadhyay M, Kodela R, Nath N, Barsegian A, Boring D, Kashfi K. Hydrogen sulfide-releasing aspirin suppresses NF-kappaB signaling in estrogen receptor negative breast cancer cells in vitro and in vivo. Biochem Pharmacol. 2012;83:723–32.
Bai YW, Ye MJ, Yang DL, Yu MP, Zhou CF, Shen T. Hydrogen sulfide attenuates paraquat-induced epithelial-mesenchymal transition of human alveolar epithelial cells through regulating transforming growth factor-beta1/Smad2/3 signaling pathway. J Appl Toxicol. 2019;39:432–40.
Li D, Yang Z, Liu Z, Zou Q, Yuan Y. Clinical significance of CBS and CCL21 in gallbladder adenocarcinomas and squamous cell/adenosquamous carcinomas. Appl Immunohistochem Mol Morphol. 2020;28:103–10.
Wang YH, Huang JT, Chen WL, Wang RH, Kao MC, Pan YR, et al. Dysregulation of cystathionine gamma-lyase promotes prostate cancer progression and metastasis. EMBO Rep. 2019;20:e45986.
Hasegawa U, Tateishi N, Uyama H, van der Vlies AJ. Hydrolysis-sensitive dithiolethione prodrug micelles. Macromol Biosci. 2015;15:1512–22.
Aiello NM, Kang Y. Context-dependent EMT programs in cancer metastasis. J Exp Med. 2019;216:1016–26.
Pastushenko I, Blanpain C. EMT transition states during tumor progression and metastasis. Trends Cell Biol. 2019;29:212–26.
Tulchinsky E, Demidov O, Kriajevska M, Barlev NA, Imyanitov E. EMT: a mechanism for escape from EGFR-targeted therapy in lung cancer. Biochim Biophys Acta Rev Cancer. 2019;1871:29–39.
He X, Xue M, Jiang S, Li W, Yu J, Xiang S. Fucoidan promotes apoptosis and inhibits EMT of breast cancer cells. Biol Pharm Bull. 2019;42:442–7.
Cho ES, Kang HE, Kim NH, Yook JI. Therapeutic implications of cancer epithelial-mesenchymal transition (EMT). Arch Pharmacol Res. 2019;42:14–24.
Liao CC, Chen YH, Lin F, Qi YF. Hydrogen sulfide inhibits transforming growth factor beta-1 induced bronchial epithelial-mesenchymal transition. Chin Med J (Engl). 2015;128:3247–50.
Miyazaki T, Kato H, Nakajima M, Sohda M, Fukai Y, Masuda N, et al. FAK overexpression is correlated with tumour invasiveness and lymph node metastasis in oesophageal squamous cell carcinoma. Br J Cancer. 2003;89:140–5.
Zaidel-Bar R, Itzkovitz S, Ma’ayan A, Iyengar R, Geiger B. Functional atlas of the integrin adhesome. Nat Cell Biol. 2007;9:858–67.
Lahlou H, Sanguin-Gendreau V, Zuo D, Cardiff RD, McLean GW, Frame MC, et al. Mammary epithelial-specific disruption of the focal adhesion kinase blocks mammary tumor progression. Proc Natl Acad Sci USA. 2007;104:20302–7.
Yu HG, Nam JO, Miller NL, Tanjoni I, Walsh C, Shi L, et al. p190RhoGEF (Rgnef) promotes colon carcinoma tumor progression via interaction with focal adhesion kinase. Cancer Res. 2011;71:360–70.
Deakin NO, Turner CE. Distinct roles for paxillin and Hic-5 in regulating breast cancer cell morphology, invasion, and metastasis. Mol Biol Cell. 2011;22:327–41.
Fang LP, Lin Q, Tang CS, Liu XM. Hydrogen sulfide attenuates epithelial-mesenchymal transition of human alveolar epithelial cells. Pharmacol Res. 2010;61:298–305.
Cheng S, Lu Y, Li Y, Gao L, Shen H, Song K. Hydrogen sulfide inhibits epithelial-mesenchymal transition in peritoneal mesothelial cells. Sci Rep. 2018;8:5863.
Acknowledgements
This study was supported by grants from the Chinese National Natural Sciences Foundation (81773099, 81630092 and 82130106), Department of Science and Technology of Jiangsu Province (BK20192005) and the program B for Outstanding PhD candidate of Nanjing University (202001B051).
Author information
Authors and Affiliations
Contributions
ZCH, HQZ and JC (Jian Cheng) designed the outline of the paper. HQZ revised this paper. FFC and HRX contributed equally to this work. FFC and HRX performed most of the experiments in this study. FFC wrote the paper and prepared the Figures. SHY, PL, YYL, JC (Jia Chen), ZQB and HSS helped with the cell related experiments. SHY and PL helped with the experiments using animals. All authors have read and approved the final version of this paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
Animal welfare and experimental procedures were performed in strict accordance with high standard animal welfare and other related ethical regulations approved by the Nanjing University Animal Care and Use Committee.
Supplementary information
Rights and permissions
About this article

Cite this article
Cai, Ff., Xu, Hr., Yu, Sh. et al. ADT-OH inhibits malignant melanoma metastasis in mice via suppressing CSE/CBS and FAK/Paxillin signaling pathway. Acta Pharmacol Sin 43, 1829–1842 (2022). https://doi.org/10.1038/s41401-021-00799-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00799-x
