
Abstract
Staurosporine, together with such examples as penicillin, aspirin, ivermectin and sildenafil, exemplifies the role that serendipity has in drug discovery and why 'finding things without actually searching for them' retains a prominent role in drug discovery. Hitherto not clinically useful, due to its potency and promiscuity, new delivery technology is opening up new horizons for what was previously just the parent compound of innovative, highly-successful anti-cancer agents.
Similar content being viewed by others

Staurosporine is a naturally occurring, extraordinary chemical produced by a soil-dwelling microbe. It revolutionized the field of anti-cancer therapy and is now itself offering the prospect of yet further advances in human health, as well as in the control of a variety of pests and parasites. However, it is illustrative to examine the pathway by which it was discovered to help provide an influential evidence base for future drug discovery.
In Nature, microorganisms do not produce meaningless or futile chemicals, they all have a purpose. In essence, it is simply that we have yet to discover most of them or identify their true usefulness for humans. Historically, beneficial human medications were discovered via trial and error and, eventually, by scientific identification of the active ingredient in traditional remedies. Crude extracts or purified chemicals, mostly from natural sources, began to be screened for biological activity without consideration for any specific biological target. In other words, ‘Bioactivity first—Chemical compound second’. However, in the late-1960s, when lead compounds for antibiotic development were becoming increasingly scarce, the Ōmura research group at Tokyo’s Kitasato Institute envisaged that an innovative ‘New way of looking’ was required. Consequently, we embraced a new philosophy to govern the search for useful chemicals from microbial origins, namely a ‘Compound first—Bioactivity second’ approach [1]. This concept was exemplified by introduction of a novel Physico-chemical screening programme, which facilitated discovery of a diverse range of useful compounds whose important bioactivity was often identified several years after discovery (Fig. 1) [2]. The approach also helped overcome the challenge of exploiting microbes that proved to be difficult to cultivate using standard practices (99% of the total). Over the last couple of decades, drug discovery research has evolved into a classical targeted pharmacology approach, which now encompasses screening of entire chemical libraries, in cells or whole organisms, to identify compounds that have a desirable and specific therapeutic effect.

Physico-chemical screening discovery & key bioactivity identification. a Pyrindicin was shown to possess weak anti-microbial and several pharmacological activities. b NA-337A possessed fat clearing properties. c TM-64 induced weak deteriorated reflex action of cornea in guinea pigs. d Quinoline-2-methanol affected hypoglycaemic activity in rats. e Dityromycin interacted with ribosomal protein S12 on small subunits to display anti-microbial activity. f Staurosporine inhibits protein kinases. g Herquline A inhibits platelet aggregation. h Neoxaline inhibits tubulin polymerization. i Reductinomycin exhibits anti-tumour activity against Ehrlich ascites carcinoma in mice. j Sespendole inhibits mouse macrophage synthesis of cholesteryl ester and triacylglycerol. k Spoxazomycin exhibits anti-trypanosomal activity against Trypanosoma brucei brucei
Traditionally, plants formed the basis of human medicine practiced for thousands of years, and remain highly valued worldwide as a rich source of therapeutic agents for the treatment and prevention of diseases and ailments. Over 35,000 plant species are used for medicinal purposes globally [3]. Following the 1928 discovery of penicillin, terrestrial microorganisms quickly began to be exploited as a virtually endless source of structurally diverse bioactive substances. In Western medicine today, >50% of pharmaceutical commodities contain natural products or are synthesized from them, with 10–25% of all prescription drugs containing one or more plant-derived ingredients [4]. It has been reported that around 80% of current antimicrobial, cardiovascular, immunosuppressive and anti-cancer drugs come from plant origins [5]. Increasingly over the past century, microorganisms have become the primary source of such therapeutic agents, so far yielding a broad spectrum of highly successful antibiotic agents, including penicillins, cephalosporins, aminoglycosides, tetracyclines and macrolides.
Alkaloids retain a leading place in that assemblage, being produced by a large variety of organisms, including bacteria, fungi, animals and especially higher plants, 10–25% of which contain alkaloids. Alkaloids are a group of diverse biomolecules, all secondary compounds derived from amino acids or via the transamination process. These compounds have been widely used in traditional or modern medicine or as starting points for drug discovery. They are a unique group of chemicals, active at different cellular levels, and critical for fundamental biological processes of plants, animals and microorganisms. Most known properties of alkaloids are fundamental to survival. Plant alkaloids are essentially involved in protection or growth regulation. In animals, alkaloid-related substances such as serotonin, dopamine and histamine are important neurotransmitters. Alkaloids are biotoxic, but not to the organisms that produce them, their toxicity being directed towards foreign organisms or cells in which they can alter DNA and selectively disrupt cells. Moreover, they play a very important role in the immune systems of animals and plants. In biomedicine, alkaloids have an extensive range of pharmacological properties, including analgaesic, anti-asthma, anti-cancer, anti-arrhythmic, anti-bacterial, anti-hyperglycaemic, anti-malarial, cholinomimetic and vasodilatory. The centuries-old widespread therapeutic use of alkaloid-containing plants meant that when the first alkaloids were isolated in the 19th century, they were immediately exploited in clinical practice [6]. Consequently, the Kitasato research group concluded that searching for alkaloids of microbial origin promised a panoply of useful novel bioactive ingredients with drug-like properties. That approach has increasingly proved to be both sound and productive. The search for new therapeutic agents from natural sources has been rapidly intensifying over the past 40 years, leading to the accumulation of a remarkably diverse array of over 139,000 natural products [7]. All these compounds are potential candidates for drug development. As an example of the impact, between 1981–2006 in the North American, European, and Japanese markets, 47.1% of a total of 155 clinically-approved anti-cancer drugs were derived from Nature [8], with staurosporine being a key compound in this respect.
In the late-1960s, the Ōmura group at the Kitasato Institute decided to proactively pursue the new ‘Compound first—Bioactivity second’ approach. The team introduced an innovative Physico-chemical Screening method, initially using Dragendorff’s reagent, as part of a new search specifically for alkaloids from naturally occurring microorganisms. The goal was to identify and isolate alkaloid compounds and subsequently to make them available for screening, either by us or by other research groups. As one of the findings, in 1976 the group discovered the world’s first indolocarbazole compound [9]. Isolated from a soil-dwelling microorganism, Streptomyces staurosporeus (now Lentzea albida), we code-named the compound AM-2282, later giving it the name ‘staurosporine’ (Fig. 2.). At the time, none of us could ever have imagined that staurosporine would turn out to be the forerunner of a new class of novel drugs that would go on to revolutionize cancer treatment. Nor that it would become a globally leading chemical reagent and outstanding drug lead compound, attracting global attention from chemists, biologists, physicians and the pharmaceutical industry [10]. Or that its widespread use and impact would help emphasize that committed research collaborations between academia and industry can create and expedite enormous progress and substantial improvements in public health and welfare worldwide.

Staurosporine producing organism. Scanning electron micrograph of Lentzea albida AM-2282. Scale bar: 1 µm
Staurosporine was extracted from a culture of a soil sample collected in Iwate Prefecture, Japan and its exact molecular structure determined later in 1994 by X-ray crystallography [11]. Our initial assays demonstrated that staurosporine possessed promising anti-fungal and hypotensive properties but lacked any anti-bacterial bioactivity [9]. Subsequent research reported that the compound also demonstrated platelet aggregation inhibition [12] and anti-hypertensive properties [13]. Of much greater significance, a decade after we discovered the compound, another group of researchers made the breakthrough observation that staurosporine was an extremely potent but non-specific inhibitor of protein kinases, particularly tyrosine kinases, and that it had a remarkably strong cytotoxic effect on cancer cells. In 1986, the isolation and complete structure elucidation of K252a, a biosynthetic precursor of staurosporine, was announced, with the compound proving to be a potent protein kinase C (PKC) inhibitor, with an IC50 of 32 nM [14]. The same year it was reported that staurosporine itself also inhibited PKC but with a slightly higher affinity (IC50 = 2.7 nM) [15]. This discovery heralded enormous potential for using staurosporine as an anti-cancer agent. More than 90 tyrosine kinases are known to be critical for malignant transformation and tumour angiogenesis [16]. Tyrosine kinase inhibitors (TKIs), which can target both receptor and cytoplasmic kinases, can improve cancer outcomes by controlling the activation of kinases in cancer cells [17, 18]. However, although the compound remains one of the most potent inhibitors ever found, it proved to be extremely promiscuous, interacting with many other kinases (over 250 known to date), including those in blood plasma. Interaction with kinases with a Kd less than 3 μM [19] precluded its therapeutic use. Nevertheless, it served as a unique, invaluable starting point for implementing function-oriented synthesis strategies [20], stimulating a comprehensive 20-year flow of research reports (Fig. 3.).

Staurosporine research articles from 1977–2014. Data source is SciFinder® (Chemical Abstracts Service)
Over the last 30 years, indolocarbazole compounds have been isolated from a variety of organisms, including bacteria, fungi and invertebrates. Post-1986, development of small-molecule kinase inhibitors quickly became one of the most widely and intensely pursued areas of drug discovery worldwide, especially for combatting cancer via targeted therapy. The unusual architecture of the indolocarbazoles, coupled with their excellent biological activity, led many researchers to attempt to synthesize it, culminating in the first total synthesis of staurosporine reported by the Danishefsky and Wood groups in 1996 [21, 22].
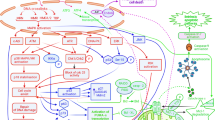
Protein kinases regulate essential aspects of cells, including metabolism, cell cycle progression and cytoskeletal arrangement. They catalyze the reversible transfer of the γ-phosphate group of adenosine triphosphate (ATP) onto a substrate, mediating signal transduction and thereby regulating cellular processes, including cell proliferation, survival, apoptosis, metabolism, transcription and differentiation, as well as other systems (Fig. 4.) [23, 24]. It was hypothesized that a vertebrate genome could encode more than 1,000 protein kinases but an analysis of the human genome published at the turn of the century identified only 518 protein kinase genes [25]. The actual number of protein kinases in the human genome remains a matter of conjecture, with Kinasenet (http://www.kinasenet.ca) providing information on over 530 and other researchers reporting that there are over 600 putative kinase genes in the human genome, some 3% of all human protein-coding genes [26]. All catalytic domains of the kinases share homologous structures for binding ATP, binding which occurs at varying degrees of strength and specificity depending on the compound. Pharmacological and pathological evidence has confirmed that kinases are promising drug targets for tackling not just cancer but a broad spectrum of diseases [27,28,29], including inflammatory conditions [30, 31], central nervous system (CNS) disorders [32], cardiovascular diseases [33] and diabetes [34].

Model of signal transduction and staurosporine inhibition. ANP, Atrial natriuretic peptide; R, Receptors; PIP2, Phosphatidylinositol 4,5-bisphosphate; IP3, Inositol triphosphate; DG, Diacylglycerol; SR, Sarcoplasmic reticulum; CaM, Calmodulin; GC, Guanylate cyclase; AC, Adenylate cyclase; G, G protein-coupled receptors; cGMP, Cyclic-3′,5′-guanosine monophosphate; cAMP, Cyclic-3′,5′-adenosine monophosphate; PKC, Protein kinase C; MLCK, Myosin light chain kinase; PKG, cGMP-dependent protein kinase; PKA, cAMP-dependent protein kinase; GTP, Guanosine triphosphate; ATP, Adenosine triphosphate; P, Phosphorylation
The degree of specificity, or lack of it, has traditionally been the major obstacle to kinase drug discovery. But that appears to be changing. Still one of the most potent inhibitors found, the lack of specificity shown by staurosporine has, to date, prevented it from being used for clinical purposes. But it has had a paramount and unique role as the parent compound for a variety of highly successful derivatives and analogues. Initial research on staurosporine led to identification and development of the pharmacophore model which, in turn, led to the synthesis of kinase inhibitors with greater specificity, effective against tyrosine kinases, PKC, cyclin-dependent kinases and G-protein-coupled receptor kinases (Fig. 5). The new targetted therapy approach produced drugs having a higher specificity towards tumour cells, usually with less toxicity. The end product, rationally-designed kinase inhibitors also proved to be useful in combination with traditional indiscriminate cytotoxic chemotherapy or radiation treatment to produce an overall synergistic, complementary and significantly improved anti-cancer effect [35].

History of protein kinase inhibitor development. Src, Proto-oncogene tyrosine-protein kinase; BCR-ABL, Fusion gene of Bcr gene on chromosome 22 and the c-Abl gene on chromosome 9; p38MAPK, p38 mitogen-activated protein kinase; FGFR, Fibroblast growth factor receptor; EGFR, Epidermal growth factor receptor; PDGF, Platelet derived growth factor; VEGF, Vascular endothelial growth factor; CDKs, Cyclin-dependent kinases; PKA, Protein kinase A; HA1077 (Erll®), Vasodilative agent; Imatinib (Gleevec®), BCR-ABL inhibitor; EML4-ALK, Fusion gene of echinoderm microtubule associated protein-like 4 and anaplastic lymphoma kinase; Crizotinib (Xalkori®), EML4-ALK inhibitor; Midostaurin (Rydapt®), Multikinase inhibitor
Since the mid-1980s, protein kinases have been the primary cellular targets with respect to anti-cancer agents, becoming the second most important group of drug targets, after G-protein-coupled receptors [36], with kinases in general and tyrosine kinases in particular accounting for almost half of all newly approved anti-cancer drugs [37]. The ground-breaking approval by the US Food and Drug Administration (FDA) of the first kinase inhibitor, imatinib—which is derived directly from staurosporine via a phenylaminopyrimidine derivative (Fig. 6.)—occurred in 2001. Fortunately, this inaugural protein kinase inhibitor, released onto the market under the Novartis trade name Gleevec/Glivec®, proved to be extremely selective, well tolerated by patients and with few side effects [38,39,40]. It revolutionized the treatment of chronic myeloid leukaemia (CML), a cancer of white blood cells, reducing it from being a potentially fatal cancer to the status of a chronic disease. Imatinib changed the prognosis for CML so dramatically that patients diagnosed early and starting imatinib treatment have a normal life expectancy [41, 42] compared with a historical average survival time of 2 to 3 years. Imatinib was followed by a steady stream of approvals for similar compounds throughout the first decade of this century, almost one new approval annually on average. Thereafter, a cascade of new approvals has occurred over the last five years, the FDA approving a total of 37 small molecule kinase inhibitors by 2017 (Table 1 and Supplemental Figure 1.). This represents an unmatched achievement in the history of pharmaceutical research.

Gleevec® derivation from staurosporine. CGP-52411, CGP-52411 is a selective inhibitor of EGFR. CGP-57148, CGP-57148 is a compound of the 2-phenylaminopyrimidine class that selectively inhibits activity of the ABL 1(Abelson murine lukaemia viral oncogene homologue 1) and PDGFR (Platelet derived growth factor) protein tyrosine kinases
Among other staurosporine derivatives (Fig. 7.), UCN-01 (7-hydroxystaurosporine), which is identical to a natural product, inhibits several protein kinases and is currently in clinical trials against leukaemias, lymphomas, advanced solid tumours, melanomas and small-cell lung cancer. It also enhances the cytotoxicity of other anti-cancer drugs [43]. Lestaurtinib, inhibits autophosphorylation and signalling of neurotrophin-specific Trk receptors and displays marked anti-tumour activity [44]. Midostaurin, a semi-synthetic derivative of staurosporine, is also a potent but non-specific protein kinase inhibitor, notably of PKC, VEGF and FLT3, preventing tumour angiogenesis and cell proliferation, and is currently being tested against acute myeloid leukaemia [45].

Chemical structures of staurosporine derivatives. a staurosporine, b UCN-01, c midostaurin, d staurosporine lactone form, e rebeccamycin, f becatecarin, g edotecarin, h K252a, i lestaurtinib, j CEP-1347, k ruboxistaurin, l enzastaurin, m NGIC-I
Currently, >3,000 compounds, active against a wide range of protein kinases, are being investigated pre-clinically for various cancers, ophthalmic diseases, central nervous system disorders, osteoporosis and other ailments, with >130 novel TKIs undergoing clinical trials [46]. As an indication of the enormous focus on these compounds, both in industry and academia, over 1 million kinase research papers have been published, >5,000 crystal structures of kinases have been identified, plus inhibition assays have been developed for over 80% of the human kinome. Around one-third of all protein targets currently under investigation in the pharmaceutical industry are either protein or lipid kinases [47] and kinase inhibitors now make up a major portion of all newly-approved drugs [40]. Nevertheless, so far, small-molecule kinase inhibitors have been identified for only 20–30% of the human kinome.
Among the clinically approved kinase inhibitors (Fig. 8.), most are tyrosine kinase inhibitors (TKIs) [48], a handful are serine/threonine kinase inhibitors, and idelalisib and copanlisib, are phosphoinositide 3-kinase inhibitors (Fig. 8f). The molecular structures of the various TKIs are shown in Fig. 8a–f. The majority of TKIs are promiscuous, inhibiting 10–100 off-target kinases, with varying degrees of potency [49, 50]. Most are reversible inhibitors, only five, afatinib, ibrutinib, osmertinib, neratinib and acalabrutinib, are irreversible. The irreversible inhibitors are expected to produce greater specificity and potency, although concerns have been raised regarding potential toxicities.



Chemical structures of approved kinase inhibitors. a Abl inhibitors, b ALK inhibitors, c Raf inhibitors, d EGFR inhibitors, e VEGFR inhibitors, f other kinase inhibitors
As an indicator of the economic impact of these drugs, worldwide sales of ibrutinib alone are forecast to reach $9 billion by 2020 [51], with the global inhibitors market estimated to be worth $105 billion. In 2014, sales of small molecule kinase inhibitors generated around $18.5 billion, while the global market for protein kinase inhibitors is forecast to grow to $31.2 billion by 2019, with further expansion to 2025 [52,53,54]. If current trends continue, up to 50 kinase inhibitors could be in clinical use by the end of the decade, including new rationally designed drugs [40].
The pharmaceutical industry spends some $135 billion annually on R&D and has been the main driving force in drug discovery and development in pursuit of potential profit. Academia has been responsible for advancing knowledge, fundamentals and understanding of diseases, pathology and biomedical mechanisms, identifying relevant biochemical targets in the process. The success of Gleevec® reinforced the value of good target ID, target validation, hit ID, lead optimization and accurate pre-clinical assessment. Furthermore, it re-emphasized the essential role of collaboration and committed partnerships between academia (University of Pennsylvania/University of Chicago/Oregon Health & Science University) and industry (Novartis), which had been demonstrated earlier by the research collaboration that had been established between the Kitasato Institute and Merck and Co. Inc. which saw the discovery and development of ivermectin. The immeasurable value of such partnerships is emphasized by the recent Accelerating Medicines Partnership (AMP), a $230 million initiative set up between the US’s National Institutes of Health (NIH), 10 major pharmaceutical companies and several Non-Governmental Organisations (NGOs) working on specific diseases, to advance and accelerate drug discovery projects, focussing on Alzheimer’s disease, Type-2 diabetes and two autoimmune diseases, lupus and rheumatoid arthritis, the approval of tofacitinib in 2012 having established the concept for a new treatment for arthritis.
Natural products, such as staurosporine, which usually contain pharmacophores and scaffolds that differ from most synthetic kinase inhibitors, are a useful source to inspire the synthesis of novel compounds and subsequent construction of libraries with expanded structural diversity. This was clearly demonstrated by imatinib, with its origins in staurosporine, which has long been the subject of various Structure/Activity Relationship (SAR) studies to guide the design of next-generation inhibitors and provide a deeper understanding of the inhibition mechanism.
Ground-breaking understanding of cellular signalling cascades at the molecular level has led to major advances in kinase research over the past few decades. During that period, the constant challenge has been the daunting task of developing kinase inhibitors with potent inhibition against desirable targets and minimal interactions with accidental targets. Initially, selective kinase inhibitors were actively pursued. However, recent thinking has switched to adopt a theory that inhibitors with favourable selectivity or multi-target selectivity might be more suitable for cancer treatment. It has become clear that kinase inhibitors do not have to be absolutely selective; preferably, a good selectivity profile is needed to balance efficacy and toxicity. The traditional Western approach to medicine has been a reductionist one, with industry and regulators focussing predominantly on a single target. Yet some of the most historically successful drugs, such as aspirin, serotonin re-uptake inhibitors, as well as imatinib, all act on multiple targets, effectively meaning a single drug treats several diseases, a circumstance defined as ‘Polypharmacology’ [55]. Industry has usually been driven to develop drugs that have singular specificity and high affinity but there is little evidence that either is a prerequisite for either safety or efficacy. Recently, the goal of drug design appears to have been rapidly changing to the 'one drug, multiple targets' concept, such that Polypharmacology is fast becoming the new paradigm in drug discovery. This will expedite identification of drug targets, including 'secondary targets', as well as accelerating the discovery of new applications for, and re-purposing of, existing molecules [56].
All currently used TKIs are administered orally, with many having a lengthy elimination half-life. Although staurosporine is the most potent protein kinase inhibitor so far discovered, persistent issues of pharmacokinetics, toxicity and non-specificity have prevented it from being safely delivered to tumours, meaning it cannot be used in cancer treatment. However, recent innovative drug re-formulation initiatives open up the prospect of the drug being used clinically. Liposome-based delivery mechanisms have been employed to enhance drug efficacy and lower toxicity. A novel process has been developed to encapsulate staurosporine in PEG liposome nanoparticles efficiently, with a favourable drug release profile. When injected into a mouse gliobastoma model, liposomal staurosporine accumulated in tumours and suppressed them with no apparent side effects [57]. This has since been refined to achieve a 100% drug loading efficiency within 15 min of incubation at a drug-to-lipid ratio of 0.31 (mol) via an ammonium gradient. The staurosporine nanoparticles proved stable in storage and in the presence of serum. Consequently, compared with free staurosporine, a 3-fold higher dose could be delivered and safely tolerated by BALB/c mice, resulting in almost complete growth inhibition of multidrug-resistant breast tumours, while less concentrated free staurosporine only exhibited moderate activity, the researchers concluding that the new delivery method could be used for effective cancer treatment [58].
Recently, staurosporine has also begun to show promise in tackling parasites, with parasite signalling pathways now attracting increasing attention as possible drug targets. Protein kinases are essential in the growth and proliferation of malarial parasites [59] as well as tuberculosis mycobacteria [60]. Apoptosis in single-cell parasites, such as Trypanosoma and Leishmania, has been increasingly examined [61,62,63,64]. Staurosporine has been shown to induce cell death in T. brucei [65] and genetic and biochemical research has identified critical roles for protein kinases in the growth and infectivity of trypanosomatid parasites, with substantial drug development programmes now focussing on staurosporine-class anti-parasitic drug development [55,56,57,58,59, 66,67,68,69,70]. The LEISHDRUG Consortium is employing a multidisciplinary approach to reveal Leishmania kinases associated with parasite-specific pathways that can be exploited to expedite anti-leishmanial drug development.
In addition, it has recently been reported that staurosporine possesses insecticidal properties, inducing apoptosis in lepidopteran Sf9 cell lines, the authors suggesting that the compound could have potential as an insecticide against lepidopteran agricultural pests [71].
In fungi, kinases regulate signalling governing drug resistance, stress adaptation and pathogenesis. Currently, research is being concentrated on the opportunistic fungal pathogen, Candida albicans, a leading cause of morbidity and mortality in immunocompromised humans, killing around 40% of people with systemic bloodstream infection. Globally, candidiasis is one of the most frequent hospital acquired infections, with around 60,000 cases of systemic candidiasis annually, costing an estimated $2–4 billion, in the USA alone [72]. Staurosporine is capable of evading or negating fungal drug resistance, thereby expanding the range of chemical scaffolds affecting drug resistance and virulence traits. It potentiates the efficacy of azoles and echinocandins via inhibition of Pkc1 [73] and induces fungal morphogenesis via a mechanism that is independent of Pkc1 but which involves adenyl cyclase Cyr1 and the cyclic AMP-dependent protein kinase A (cAMP-PKA). The cAMP-PKA signalling cascade is critical for morphogenesis and EFG1 is an important regulator for the switch from yeast like cells to filamentous cells, that transition being recognised as one of the key factors in the virulence of C. albicans [74, 75]. Researchers conclude that staurosporine can not only improve understanding of fungal virulence mechanisms but can also be a useful compound in the development of novel, customised anti-fungal compounds [76].
Having been the chemical origin of the revolutionary change away from indiscriminate cell death arising through cytotoxic cancer therapy towards a more targeted apoptosis approach, it is eminently possible that staurosporine may continue to be an increasingly innovative and useful chemical. Continuing research on this multifaceted compound is indicating increasingly beneficial and diverse uses for this remarkable chemical, in biomedical, agricultural and other applications.
References
Ōmura S. Microbial metabolites: 45 years of wandering, wondering and discovering. Tetrahedron. 2011;67:6420–59.
Ōmura S. Splendid gifts from microorganisms. Kitasato Institute for Life Sciences. Tokyo: Kitasato University; 2015.
Yirga G, Teferi M, Kasaye M. Survey of medicinal plants used to treat human ailments in Hawzen district, Northern Ethiopia. Int J Biodivers Conserv. 2011;3:709–14.
Cameron SI, Smith RF, Kierstead KE. Linking medicinal/nutraceutical products research with commercialization. Pharm Biol. 2005;43:425–33.
Pan S-Y, et al. New perspectives on how to discover drugs from herbal medicines: CAM’s outstanding contribution to modern therapeutics. Evid Based Complement Altern Med. 2013;2013:627375.
Aniszewski T. Alkaloids—secrets of life. 129. Amsterdam: Elsevier; 2007.
Sithranga Boopathy N, Kathiresan K. Anticancer drugs from marine flora: an overview. J Oncol. 2010;2010:214186.
Newman DJ, Cragg GM. Natural products as sources of new drugs over the last 25 years. J Nat Prod. 2007;70:461–77.
Ōmura S, et al. A new alkaloid AM-2282 of Streptomyces origin. Taxonomy, fermentation, isolation and preliminary characterization. J Antibiot. 1977;30:275–82.
Sánchez C, Méndez C, Salas JA. Indolocarbazole natural products: occurrence, biosynthesis, and biological activity. Nat Prod Rep. 2006;23:1007–45.
Funato N, et al. Absolute configuration of Staurosporine by X-ray analysis. Tetrahedron Lett. 1994;35:1251–4.
Schächtele C, Seifert R, Osswald H. Stimulus-dependent inhibition of platelet aggregation by the protein kinase C inhibitors polymyxin B, H-7 and staurosporine. Biochem Biophys Res Commun. 1988;151:542–7.
Hachisu M, Hiranuma T, Koyama M, Sezaki M. Antihypertensive compounds with potent protein kinases inhibitory activity. Life Sci. 1989;44:1351–62.
Kase H, Iwahashi K, Matsuda Y. K-252a, a potent inhibitor of protein kinase C from microbial origin. J Antibiot. 1986;39:1059–65.
Tamaoki T, et al. Staurosporine, a potent inhibitor of phospholipid/Ca++ dependent protein kinase. Biochem Biophys Res Commun. 1986;135:397–402.
Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005;353:172–87.
Steeghs N, Nortier JW, Gelderblom H. Small molecule tyrosine kinase inhibitors in the treatment of solid tumors: an update of recent developments. Ann Surg Oncol. 2007;14:942–53.
Chen MH, Kerkelä R, Force T. Mechanisms of cardiac dysfunction associated with tyrosine kinase inhibitor cancer therapeutics. Circulation. 2008;118:84–95.
Karaman MW, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–32.
Wender PA, et al. Function through bio-inspired, synthesis-informed design: step economical syntheses of designed kinase inhibitors. Org Chem Front. 2014;1:1166–71.
Link JT, et al. Staurosporine and ent-staurosporine: the first total synthesis, prospects for a regioselective approach, and activity profiles. J Am Chem Soc. 1996;118:2825–42.
Wood JL, Stoltz BM, Goodman SN. Total synthesis of (+)-RK-286c, (+)-MLR-52, (+)-staurosporine, and (+)-K252a. J Am Chem Soc. 1996;118:10656–7.
Johnson LN, Lewis RJ. Structural basis for control by phosphorylation. Chem Rev. 2001;101:2209–42.
Adams JA. Kinetic and catalytic mechanisms of protein kinases. Chem Rev. 2001;101:2271–90.
Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–34.
Milanesi L, et al. Systematic analysis of human kinase genes: a large number of genes and alternative splicing events result in functional and structural diversity. BMC Bioinform. 2005;6(Suppl 4):S20.
Rask-Andersen M, Zhang J, Fabbro D, Schiöth HB. Advances in kinase targeting: current clinical use and clinical trials. Trends Pharmacol Sci. 2014;35:604–20.
Huang M, Shen A, Ding J, Geng M. Molecularly targeted cancer therapy: some lessons from the past decade. Trends Pharmacol Sci. 2014;35:41–50.
Ma WW, Adjei AA. Novel agents on the horizon for cancer therapy. CA Cancer J Clin. 2009;59:111–37.
Clark JD, Flanagan ME, Telliez JB. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. J Med Chem. 2014;57:5023–38.
Barnes PJ. New anti-inflammatory targets for chronic obstructive pulmonary disease. Nat Rev Drug Discov. 2013;12:543–59.
Muth F, et al. Tetra-substituted pyridinylimidazoles as dual inhibitors of p38α mitogen-activated protein kinase and c-Jun N-terminal kinase 3 for potential treatment of neurodegenerative diseases. J Med Chem. 2015;58:443–56.
Kikuchi R, et al. An antiangiogenic isoform of VEGF-A contributes to impaired vascularization in peripheral artery disease. Nat Med. 2014;20:1464–71.
Banks AS, et al. An ERK/Cdk5 axis controls the diabetogenic actions of PPARγ. Nature. 2015;517:391–5.
Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther. 2005;315:971–9.
Cohen P. Protein kinases—the major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002;1:309–15.
Kinch MS. An analysis of FDA-approved drugs for oncology. Drug Discov Today. 2014;19:1831–5.
Hantschel O. Unexpected off-targets and paradoxical pathway activation by kinase inhibitors. ACS Chem Biol. 2015;10:234–45.
Rix U, et al. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood. 2007;110:4055–63.
Druker BJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–7.
Viganò I, et al. First-line treatment of 102 chronic myeloid leukemia patients with imatinib: a long-term single institution analysis. Am J Hematol. 2014;89:E184–E187.
Hehlmann R, et al. Deep molecular response is reached by the majority of patients treated with imatinib, predicts survival, and is achieved more quickly by optimized high-dose imatinib: results from the randomized CML-study IV. J Clin Oncol. 2014;32:415–23.
Akinaga S, Sugiyama K, Akiyama T. UCN-01 (7-hydroxystaurosporine) and other indolocarbazole compounds: a new generation of anti-cancer agents for the new century? Anticancer Drug Des. 2000;15:43–52.
Shabbir M, Stuart R. Lestaurtinib, a multitargeted tyrosine kinase inhibitor: from bench to bedside. Expert Opin Investig Drugs. 2010;19:427–36.
Hatzimichael E, Georgiou G, Benetatos L, Briasoulis E. Gene mutations and molecularly targeted therapies in acute myeloid leukemia. Am J Blood Res. 2013;3:29–51.
Neul C, et al. Impact of membrane drug transporters on resistance to small-molecule tyrosine kinase inhibitors. Trends Pharmacol Sci. 2016;37:904–32.
Fabbro D. 25 years of small molecular weight kinase inhibitors: potentials and limitations. Mol Pharmacol. 2015;87:766–75.
Levitzki A. Tyrosine kinase inhibitors: views of selectivity, sensitivity, and clinical performance. Annu Rev Pharmacol Toxicol. 2013;53:161–85.
Davis MI, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–51.
Hantschel O, Grebien F, Superti-Furga G. The growing arsenal of ATP-competitive and allosteric inhibitors of BCR-ABL. Cancer Res. 2012;72:4890–5.
Garber K. Kinase inhibitors overachieve in CLL. Nat Rev Drug Discov. 2014;13:162–4.
Pharma Reports. Kinase inhibitors for treating cancer: Industry analysis, R&D trends and world market forecasts 2014-24. London: Visiongain; 2014.
World kinase inhibitors market: Opportunities and forecasts, 2014–22. Portland: Allied Market Research; 2016.
Pharma Reports. Kinase Inhibitors for cancer treatment: World industry analysis, R&D and market forecasts 2015-25. London: Visiongain; 2015.
Boran AD, Iyengar R. Systems approaches to polypharmacology and drug discovery. Curr Opin Drug Discov Devel. 2010;13:297–309.
Reddy AS, Zhang S. Polypharmacology: drug discovery for the future. Expert Rev Clin Pharmacol. 2013;6:41–47.
Mukthavaram R, et al. High-efficiency liposomal encapsulation of a tyrosine kinase inhibitor leads to improved in vivo toxicity and tumor response profile. Int J Nanomed. 2013;8:3991–4006.
Tang WL, Chen WC, Roy A, Undzys E, Li SD. A simple and improved active loading method to efficiently encapsulate staurosporine into lipid-based nanoparticles for enhanced therapy of multidrug-resistant cancer. Pharm Res. 2016;33:1104–14.
Doerig C, Meijer L. Antimalarial drug discovery: targeting protein kinases. Expert Opin Ther Targets. 2007;1:279–90.
Fernandez P, et al. The Ser/Thr protein kinase PknB is essential for sustaining mycobacterial growth. J Bacteriol. 2006;188:7778–84.
Debrabant A, Lee N, Bertholet S, Duncan R, Nakhasi HL. Programmed cell death in trypanosomatids and other unicellular organisms. Int J Parasitol. 2003;33:257–67.
Figarella K, et al. Prostaglandin D2 induces programmed cell death in Trypanosoma brucei bloodstream form. Cell Death Differ. 2005;12:335–46.
Figarella K, et al. Prostaglandin-induced programmed cell death in Trypanosoma brucei involves oxidative stress. Cell Death Differ. 2006;13:1802–14.
Lee N, et al. Programmed cell death in the unicellular protozoan parasite Leishmania. Cell Death Differ. 2002;9:53–64.
Barth T, et al. Staurosporine-induced cell death in Trypanosoma brucei and the role of endonuclease G during apoptosis. Open J Apoptosis. 2014;3:16–31.
Doerig C. Protein kinases as targets for anti-parasitic chemotherapy. Biochim Biophys Acta. 2004;1697:155–68.
Chow C, Cloutier S, Dumas C, Chou MN, Papadopoulou B. Promastigote to amastigote differentiation of Leishmania is markedly delayed in the absence of PERK eIF2alpha kinase-dependent eIF2alpha phosphorylation. Cell Microbiol. 2011;13:1059–77.
Madeira da Silva L, Beverley SM. Expansion of the target of rapamycin (TOR) kinase family and function in Leishmania shows that TOR3 is required for acidocalcisome biogenesis and animal infectivity. Proc Natl Acad Sci USA. 2010;107:11965–70.
Dujardin JC, Gonzalez-Pacanowska D, Croft SL, Olesen OF, Spath GF. Collaborative actions in anti-trypanosomatid chemotherapy with partners from disease endemic areas. Trends Parasitol. 2010;26:395–403.
Grant KM, et al. Inhibitors of Leishmania mexicana CRK3 cyclin-dependent kinase: chemical library screen and antileishmanial activity. Antimicrob Agents Chemother. 2004;48:3033–42.
Zhang Y, et al. Staurosporine shows insecticidal activity against Mythimna separata Walker (Lepidoptera: Noctuidae) potentially via induction of apoptosis. Pestic Biochem Physiol. 2016;128:37–44.
Uppuluri, P, Khan A, Edwards JE in Candida albicans: Cellular and Molecular Biology. Current trends in Candidiasis Prasad R editors 5–23 Switzerland: Springer Nature; 2017.
LaFayette SL, et al. PKC signalling regulates drug resistance of the fungal pathogen Candida albicans via circuitry comprised of Mkc1, calcineurin, and Hsp90. PLoS Pathog. 2010;6:e1001069.
Sohn K, Urban C, Brunner H, Rupp S. EFG1 is a major regulator of cell wall dynamics in Candida albicans as revealed by DNA microarrays. Mol Microbiol. 2003;47:89–102.
Shapiro RS, Robbins N, Cowen LE. Regulatory circuitry governing fungal development, drug resistance, and disease. Microbiol Mol Biol Rev. 2011;75:213–67.
Xie JL, O’Meara TR, Polvi EJ, Robbins N, Cowen LE. Staurosporine induces filamentation in the human fungal pathogen Candida albicans via signalling through Cyr1 and protein kinase A. mSphere. 2017;2:e00056–17.
Acknowledgements
We would like to thank all fellow researchers and collaborators, too numerous to mention, who have been involved in the discovery, investigation and development of staurosporine and its derivatives.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Ōmura, S., Asami, Y. & Crump, A. Staurosporine: new lease of life for parent compound of today’s novel and highly successful anti-cancer drugs. J Antibiot 71, 688–701 (2018). https://doi.org/10.1038/s41429-018-0029-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-018-0029-z
This article is cited by
-
Computational discovery of novel FYN kinase inhibitors: a cheminformatics and machine learning-driven approach to targeted cancer and neurodegenerative therapy
Molecular Diversity (2024)
-
Indispensable role of microbes in anticancer drugs and discovery trends
Applied Microbiology and Biotechnology (2022)
-
Lactacystin: first-in-class proteasome inhibitor still excelling and an exemplar for future antibiotic research
The Journal of Antibiotics (2019)
-
Important role of a LAL regulator StaR in the staurosporine biosynthesis and high-production of Streptomyces fradiae CGMCC 4.576
Science China Life Sciences (2019)
