
Abstract
Two new meroterpenoid-type fungal metabolites, furanoaustinol (1) and 7-acetoxydehydroaustinol (2), were isolated from the ethyl acetate extract of a marine-derived fungal strain Penicillium sp. SF-5497, along with eight (3–10) known meroterpenoids. Their structures were elucidated mainly based on the analysis of their NMR (1D and 2D) and MS data. Particularly, the novel meroterpenoid, furanoaustinol (1), belonging to the austin group, was identified to possess an unprecedented hexacyclic ring system. Biological evaluation of these compounds revealed that furanoaustinol (1) weakly inhibited the activity of protein tyrosine phosphatase 1B in a dose-dependent manner with an IC50 value of 77.2 μM. In addition, 7-acetoxydehydroaustinol (2) and four other known meroterpenoids (5, 7, 9, and 10) weakly suppressed the overproduction of nitric oxide in lipopolysaccharide-challenged BV2 microglial cells with IC50 values of 61.0, 30.1, 58.3, 37.6, and 40.2 μM, respectively.
Similar content being viewed by others

Introduction
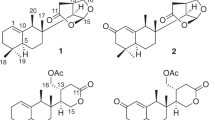
Studies on secondary metabolites produced by marine-derived fungi have recently garnered interest, mainly because of their unique structures in some cases and the fact that many of them have been reported to possess interesting pharmacological properties [1]. Meroterpenoids are distinct fungal metabolites formed by a combined terpenoid–polyketide biosynthetic pathway [2]. In the course of our ongoing search for bioactive secondary metabolites from marine-derived fungal isolates [3,4,5,6], the fungal strain, Penicillium sp. SF-5497, isolated from a sample of sea sand collected in Busan, Korea, was selected for further investigation based on the evaluation of the proton nuclear magnetic resonance (1H NMR) spectrum of the ethyl acetate (EtOAc) extract. In this study, we describe the isolation and structural elucidation of two new (1 and 2) and eight known fungal metabolites (3–10) from Penicillium sp. SF-5497 (Fig. 1). Among these, the structures and absolute configurations of the eight known metabolites, namely austinol (3) [7], austin (4) [7], austinolide (5) [8], 7-hydroxydehydroaustin (6) [9], 7-acetoxydehydroaustin (7) [7], dehydroaustin (8) [7], 11-hydroxyisoaustinone (9) [10], and 11-acetoxyisoaustinone (10) [9], were determined based on their specific rotations, mass spectrometry (MS) and NMR data, and by comparing them with the data reported in the literature.

Structures of compounds 1–10
Results and discussion
Compound 1 was obtained as a yellow gum. Its molecular formula was determined to be C25H30O9, requiring 11 unsaturations, based on the observation of an ion peak at m/z 473.1817 [M−H]− in the high-resolution electrospray ionization mass spectrometry (HRESIMS) data and the analysis of 1H and 13C NMR data. The 1H NMR spectrum (Table 1) indicated signals for terminal olefinic protons, four tertiary methyl groups, a methyl group coupled to an oxymethine group, and one oxygenated methylene signal. The 13C NMR and DEPT (distortionless enhancement by polarization transfer) spectra (Table 1) identified 25 carbon signals, including five methyl (C-12, C-14, C-15, C-9′, C-10′), four methylene (C-2, C-6, C-7, C-13), three oxymethine (C-1, C-11, C-5′), three sp3 oxygenated quaternary (C-4, C-3′, C-6′), three sp3 quaternary (C-5, C-8, C-7′), four olefinic (C-9, C-10, C-1′, C-2′), and three carbonyl carbons (C-3, C-4′, C-8′). Further, DEPT analysis and its molecular formula indicated the presence of two exchangeable protons (hydroxy groups) in 1. These data being similar to those for previously identified meroterpenoid derivatives (3–10), it was suggested that compound 1 is structurally similar to austinol (3). In the 1H NMR spectrum of 1, signals corresponding to the mutually coupled olefinic protons (H-1 and H-2) and the methyl group (H3-13) observed in 3 were absent. Instead, signals corresponding to two sp3 methylene groups (including an oxygenated one) were newly observed, suggesting that 1 and 3 structurally differ due to their modification on rings A and B. In addition, the consideration of unsaturation equivalents accounted by the analysis of its NMR data revealed that 1 possessed an additional ring in its structure compared to the ring system of 3. Comparison of the chemical shifts in the NMR data of 1 and 3, along with detailed analysis of the two-dimensional (2D) NMR data of 1 taken in dimethyl sulfoxide-d6 (DMSO-d6) and CD3OD partially confirmed the structure of 1, and provided evidence for the presence of rings A, B, and F. The spiro connection between rings A and B in 1 was established by its heteronuclear multiple bond correlation (HMBC) data that revealed heteronuclear correlations of H3-14 and H3-15 with C-5; H2-7 with C-5, C-8, and C-9; H2-2 with C-3 and C-4; and H-1 with C-3, C-4, and C-6. Comparison of the 13C chemical shifts observed for 1 with those observed for 3, and HMBC correlations of H3-12 with C-7, C-8, C-9, and C-7′, supported this connection, and allowed the chemical shift assignments of carbons involved in this partial structure. With this spiro connection between rings A and B established, HMBC correlations of the oxygenated methylene (H2-13) with the sp3 oxymethine carbon (C-1) and H2-13 with C-9 and C-10 verified the presence of tetrahydrofuran ring system (ring F) in 1 (Fig. 2). The remaining part of the molecule was deduced on the basis of HMBC correlations of H-11, H-1′, H-5′, H-9′, and H-10′ (Table 1) and comparison of the 13C chemical shifts observed in 1 with those observed in 3. Therefore, the planar structure of 1 was assigned as a novel meroterpenoid, which possesses an unprecedented hexacyclic ring system as shown.

Key COSY and HMBC correlations for furanoaustinol (1) and 7-acetoxydehydroaustin (2)
The rigid nature of 1 resulting from its highly fused hexacyclic ring system allowed assignment of its relative configuration based on the analysis of NOESY (nuclear Overhauser effect spectroscopy) data. NOESY correlations between H3-9′/H-1′a, H3-9′/H3-12, and H-1′b/H3-12 indicated that these groups are on the same face of the molecule. Further, NOESY correlations between H3-12/H3-14, H3-14/H-13α, H-2β/H3-15, and H-6α/H3-15 verified the configuration of the spiro carbon C-5 as shown. The β-configuration of H-1 was supported by NOESY correlations between H-1/H-6β and H-1/H-13β. The relative configuration of C-5′ as shown was proposed by NOESY correlations between H-5′ and H-1′b, and the pseudoaxial orientation of the hydroxy group at C-11 was determined by NOESY correlations of H-11 with H-13α and H-13β. Therefore, the relative configuration of 1 was assigned, as shown in Fig. 3.

Key NOESY correlations of furanoaustinol (1)
Compound 2 was obtained as a white gum, and the molecular formula was determined as C27H30O10 based on the observation of an ion peak at m/z 515.1914 [M+H] + in the HRESIMS data and the analysis of 1H and 13C NMR data. The 1H NMR spectrum (Table 2) displayed characteristic signals for six olefinic, five tertiary methyl, one methylene, and three oxygenated methine protons (including one coupled to a methyl group). The 13C NMR and DEPT spectra (Table 2) revealed the presence of 27 carbon signals, including 6 methyl, 1 methylene, 3 oxygenated sp3 methine, and 17 non-protonated carbons including 4 carbonyls and 2 sp2 carbons. Comparison of the 1H NMR and 13C NMR data of 2 with those of 6 indicated that most of the signals were identical, except for the chemical shift differences for two sp3 oxymethine groups (δH/δC = 4.36/76.2 and δH/δC = 5.37/67.6). Further, the identical molecular formulas of 2 and 6 suggested a regioisomerism between these compounds. After assigning chemical shift values for identical carbons and protons based on detailed analysis of the 2D NMR data, the hydroxyl and acetoxy groups in 2 were found to be on the side opposite to that in 6. Although HMBC correlation of H-7 with carbonyl carbon in the acetoxy group was absent in the HMBC data, comparison of the chemical shifts for C-7 and C-11 of compounds 2, 6, and 7 supported the assignment of planar structure of 2 as shown.
As in the case of 1, the relative configuration of 2 was elucidated based on its NOESY data. NOESY correlations, such as H3-9′/H-1′a, H3-9′/H3-12, H-1′b/H3-12, and H3-12/H3-14, observed in 2 were similar to those in 1, suggesting that 2 has the same relative configuration as that of 1 at the analogous stereogenic centers of the molecule. In addition, NOESY correlations of H3-10′/H-7 and H-1/H-7 indicated that the acetoxy group at C-7 is oriented towards the α face of the structure. Similarly, NOESY correlations of H-11 with H-13a and H-13b supported the orientation of the hydroxyl group at C-11 towards the α-face of the structure.
Our initial attempts to establish the absolute configuration of 1 using the modified Mosher’s method were unsuccessful, and more extensive efforts were hampered by sample limitations. Therefore, absolute configurations of previously defined austin analogs were included in this report. The absolute configurations of austinol (3), austin (4), 7-acetoxydehydroaustin (7), and dehydroaustin (8) have been determined by single-crystal X-ray diffraction analysis using Cu Kα radiation [11]. Since we observed positive specific rotations for these compounds, consistent with those in the literature, it was proposed that compounds 3, 4, 7, and 8 have the same absolute configurations as those defined in the literature [11]. Among the fungal meroterpenoids, austin-related meroterpenoids are referred to as 3,5-dimethylorsellinic acid (DMOA)-derived meroterpenoids because they are biosynthesized from DMOA and farnesyl pyrophosphate [10, 12]. Meroterpenoids of the austin group have been isolated from several fungal genera such as Aspergillus, Penicillium, and Emericella. Among these, the absolute configurations of the isolated meroterpenoids seem dependent on the producing organisms. For example, the (+)-form austinol has been reported from Penicillium sp. MG-11 with five other structurally closed meroterpenoids [7]. On the other hand, the (−)-form dehydroaustinol representing the antipode of austinol mentioned in this report has been isolated from Emericella dentate [13, 14]. In addition, biosynthetic studies for the austin group suggested that meroterpenoids encountered in this study are biosynthetically related [10, 12]. Although there was no positive evidence for suggesting the absolute configuration of the compounds 1, 2, 5, 6, 9, and 10 isolated in this study, the absolute configurations of these compounds was suggested to be analogous to those of compounds 3, 4, 7, and 8 based on literature survey and the assumption that the same fungus would not synthesize enantiomers of biosynthetically related compounds.
Compounds 1 and 2 possess the skeleton of DMOA-derived meroterpenoids, which cover a quite large number of structurally diverse metabolites derived from variations in the cyclization and tailing reactions with farnesylated aromatic tetraketide, DMOA [11]. Among these, meroterpenoids belonging to the austin group are considered biosynthetically unique natural products due to their rare type of spirolactone formation. Although the presence of α,β-unsaturated spirolactone ring moiety is common in these meroterpenoids, only a methyl or exocyclic olefinic group at the C-10 of position of this ring is known. In this regard, compound 1 represents a unique meroterpenoid of the austin group, which includes an additional tetrahydrofuran ring in its structure, subsequently resulting in the formation of an unprecedented hexacyclic ring system. It can be postulated that, in addition to already known biosynthetic steps for the austin group, further oxidative, reductive, and cyclization biosynthetic steps on the spirolactone moiety have been involved to form this tetrahydrofuran ring moiety.
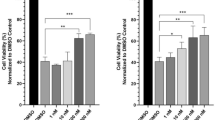
Previous studies have shown that austin (4) and related meroterpenoids, such as 7-acetoxydehydroaustin (7), are toxic to insects [7, 15]. They also selectively block the nicotinic acetylcholine receptors in cockroach [16]. Protein tyrosine phosphatase 1B (PTP1B) is involved in the negative regulation of the signaling mediated by tyrosine kinase-type receptors such as leptin and insulin receptors [17,18,19]. In addition, a number of studies proposed that PTP1B is widely expressed in cardiovascular tissues, especially in the heart and endothelium, and is involved in the manifestation of cardiovascular disease [20, 21]. Thus, PTP1B inhibitors can be applied for the treatment of cardiovascular diseases and insulin resistance such as diabetes or obesity [22]. In an assay to identify the PTP1B inhibitory effects of the isolated compounds, compound 1 was found to weakly inhibit PTP1B enzyme activity with the half-maximal inhibitory concentration (IC50) value of 77.2 μM. However, compounds 2–10 exhibited no inhibitory activity up to 100 μM. Ursolic acid was employed as a positive control showing IC50 value of 2.1 μM. In addition, the anti-inflammatory effects of the isolated compounds were also evaluated in RAW 264.7 macrophages and BV2 microglial cells. In this assay, the cells were challenged with lipopolysaccharide (LPS) to induce the production of nitric oxide (NO), a pro-inflammatory mediator, and the effects of compounds on the overproduction of NO were evaluated using the Griess method [6]. Among the tested compounds, compounds 2, 5, 7, 9 and 10 suppressed the LPS-induced overproduction of NO with IC50 values of 61.0, 30.1, 58.3, 37.6, and 40.2 μM, respectively in BV2 cells. However, none of the compounds were effective in LPS-treated RAW 264.7 cells. Butein (IC50 = 4.4 μM) was used as a positive control.
Methods
General experimental procedures
Ultraviolet (UV) spectra were recorded on a Biochrom 1300 UV/visible spectrophotometer. NMR spectra (1D and 2D) were recorded in DMSO-d6, CD3OD, or CDCl3 using a JEOL JNM ECP-400 spectrometer (400 MHz for 1H and 100 MHz for 13C) and the chemical shifts were referenced relative to the respective residual solvent signals (DMSO-d6: δH 2.50 and δC 39.5; CD3OD: δH 3.30 and δC 49.0; and CDCl3: δH 7.26 and δC 77.2). HRESIMS data were obtained using an electrospray ionization quadrupole-time of flight tandem mass spectrometry (ESI Q-TOF MS/MS) system (AB SCIEX Triple). Flash column chromatography was carried out using YMC octadecylfunctionalized silica gel (C18) column. High-performance liquid chromatography (HPLC) separations were performed on a preparative-C18 column (20 × 150 mm; 5 μm particle size) with a flow rate of 5 mL/min. Compounds were detected by UV absorption at 210 nm. Solvents used for HPLC were analytical grade.
Isolation and identification of the fungal strain
Penicillium sp. SF-5497 was isolated from a sample of sea sand collected at Gijang-gun, Busan (35 22.2257 N; 129 23.9238 E) on 5 April 2010. The sample (1 g) was mixed with sterile sea water (10 mL). An aliquot (0.1 mL) of the filtered sample was processed utilizing the spread plate method in a potato dextrose agar (PDA) medium containing sea water. The plate was incubated at 25 °C for 14 days. After sub-culturing the isolates several times, pure cultures were selected and preserved at −70 °C. The fungal strain SF-5497 was identified based on the analysis of the ribosomal RNA (rRNA) sequence. A GenBank search with the 28S rRNA sequence of SF-5497 (GenBank accession number KU341382) indicated P. brasilianum (HM469396), P. simplicissimum (HM469430), and Trematosphaeria biappendiculata (GU205229) as the closest matches showing sequence identities of 99.79, 99.64, and 99.52%, respectively. Therefore, the marine-derived fungal strain SF-5497 was characterized as Penicillium sp., but could not be definitively identified at the species level.
Fermentation, extraction, and isolation of compounds 1 and 2
Fermentation was performed in 45 Fernbach flasks, each containing 200 mL of PDA with 3% NaCl. The flasks were incubated at 25 °C for 14 days. Extraction of the combined PDA media with EtOAc (1 L each) provided an organic phase, which was then concentrated in vacuo to yield a residue of SF-5497 (7.2 g). The extract was subjected to reversed-phase (RP) C18 flash column chromatography (CC, 45 × 380 mm), and eluted with a stepwise gradient of 20, 40, 60, 80, and 100% (v/v) MeOH in H2O (500 mL each) to give fractions, SF-5497(2)-1–6. Further, fraction SF-5497(2)-4 was subjected to RP C18 flash column chromatography (CC, 45 × 380 mm), and eluted with a gradient of MeOH in H2O (50–100% v/v) to give fractions, SF-5497(2)-4-1–10. Fraction SF-5497(2)-4-3 was separated by preparative RP HPLC, and eluted with a gradient of 40–60% (v/v) MeOH in H2O (0.1% formic acid) over 50 min to give 1 (6.9 mg, tR = 29.6 min). Fraction SF-5497(2)-4-5 was subjected to silica gel CC, and eluted with a gradient of CH3Cl-MeOH to give seven fractions, SF-5497(2)-4-5-1–7. Fraction SF-5497(2)-4-5-6 was further purified using preparative RP HPLC, and eluted with a gradient of 40–60% CH3CN in H2O over 50 min to give 2 (16.1 mg, tR = 27.5 min).
Furanoaustinol (1)
Yellow gum; [α]D24 -114.8 (c 0.06, MeOH); UV (MeOH) λmax (log ε) 203 (3.12); 1H (CD3OD 400 MHz) and 13C NMR data (CD3OD, 100 MHz), see Table 1; 1H NMR (DMSO-d6, 400 MHz) δ 5.62 (1H, s, J = 1.4 Hz, H-1′a), 5.19 (1H, m, H-1′b), 4.48 (1H, q, J = 6.2 Hz, H-5′), 4.40 (1H, d, J = 13.1 Hz, H-13β), 4.32 (1H, d, J = 13.1 Hz, H-13α), 4.15 (1H, s, H-11), 3.72 (1H, dd, J = 3.2, 3.3 Hz, H-1), 3.01 (1H, dd, J = 3.2, 17.6 Hz, H-2β), 2.66 (1H, dd, J = 3.3, 17.6 Hz, H-2α), 2.14 (1H, m, H-6β), 2.11 (1H, m, H-7), 1.81 (1H, m, H-6α), 1.570 (3H, s, H-9′), 1.572 (1H, m, H-7), 1.566 (3H, s, H-12), 1.41 (3H, s, H-15), 1.31 (3H, s, H-14), 1.13 (3H, d, J = 6.07 Hz, H-10′); 13C NMR (DMSO-d6, 100 MHz) δ 172.1 (C-8′), 170.0, (C-3), 168.9 (C-4′), 144.4 (C-10), 139.0 (C-2′), 131.8 (C-9), 117.0, (C-1′), δ 86.0 (C-3′), 84.5 (C-4), δ 83.7 (C-1), 79.0 (C-6′), 78.8 (C-5′), 75.0 (C-11), 67.9 (C-13), 65.7 (C-7′), 46.7 (C-5), 38.7 (C-8), 33.2 (C-2), 32.3 (C-7), 28.6 (C-14), 27.6 (C-13), 27.4 (C-15), 26.4 (C-6), 20.9 (C-9′), 12.0 (C-10′); HMBC (DMSO-d6) H-1→C-2, C-4, C-5, C-6, H-2→C-1, C-3, H-6→C-4, C-5, C-7, C-8, H-7→C-5, C-6, C-8, H-11→C-8, C-10, C-2′, C-3′, C-9′, H-12→C-7, C-8, C-9, C-7′, H-13→C-1, C-5, C-9, C-10, H-14→C-4, C-5, C-15, H-15→C-4, C-5, C-14, H-1′→C-2′, C-3′, C-7′, H-5′→C-6′, C-10′, H-9′→C-11, C-2′, C-3′, C-4′, H-10→C-5′; HRESIMS m/z 473.1817 [M − H]− (calcd for C25H29O9, 473.1812).
7-Acetoxydehydroaustinol (2)
White gum; [α]D24 + 193.3 (c 0.17, MeOH); UV (MeOH) λmax (log ε) 204 (3.10); 1H (CDCl3, 400 MHz) and 13C NMR data (CDCl3, 100 MHz), see Table 2; HRESIMS m/z 515.1914 [M+H]+ (calcd for C27H31O10, 515.1912).
PTP1B enzyme assay
PTP1B enzyme was purchased from ATGen Co., Ltd. (Gyeonggi-do, Korea), and its activity was evaluated using p-nitro phenyl phosphate (p-NPP). The reaction mixture consisted of PTP1B (0.04 μg), 50 mM Bis-Tris (pH 6.0), 5.0 mM dithiothreitol, 2.0 mM EDTA, and 1.0 mM p-NPP, with or without the isolated compounds. After incubation at 37.5 °C for 30 min, the reaction was stopped by the addition of 10 N NaOH. Production of p-nitrophenol was evaluated by measuring the absorbance at 405 nm. Non-enzymatic hydrolysis of 1.0 mM p-NPP was compensated for by measuring the increase in absorbance at 405 nm in the absence of PTP1B enzyme
Cell culture and nitrite production determination
BV2 microglia cells were maintained at 5 × 105 cells/mL in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum, penicillin H (100 U/mL), streptomycin (100 mg/L), and l-glutamine (2 mM) and incubated at 37 °C in a humidified atmosphere containing 5% CO2. The nitrite concentration in the medium, an indicator of NO production, was measured by the Griess reaction. Each supernatant (100 μL) was mixed with an equal volume of the Griess reagent (solution A, 222488; solution B, S438081; Sigma), and the absorbance of the mixture at 540 nm was determined using an ELISA plate reader.
References
Mostafa ER, Rainer E. Secondary metabolites of fungi from marine habitats. Nat Prod Rep. 2011;28:290–344.
Simpson TJ, Ahmed SA, McIntyre CR, Scott FE, Sadler IH. Biosynthesis of polyketide-terpenoid (meroterpenoid) metabolites andibenin B and andilesin A in Aspergillus variecolor. Tetrahedron. 1997;53:4013–34.
Quang TH, Lee DS, Sohn JH, Kim YC, Oh H. A new deoxyisoaustamide derivative from the marine-derived fungus Penicillium sp. JF-72. Bull Korean Chem Soc. 2013;34:3109–12.
Lee DS, et al. Penicillinolide A: a new anti-inflammatory metabolite from the marine fungus Penicillium sp. SF-5292. Mar Drugs. 2013;11:4510–26.
Quang TH, et al. Tanzawaic acid derivatives from a marine isolate of Penicillium sp. (SF-6013) with anti-inflammatory and PTP1B inhibitory activities. Bioorg Med Chem Lett. 2014;24:5787–91.
Kim DC, et al. Dihydroisocoumarin derivatives from marine-derived fungal isolates and their anti-inflammatory effects in lipopolysaccharide-induced BV2 microglia. J Nat Prod. 2015;78:2948–55.
Hayashi H, et al. Acetoxydehydroaustin, a new bioactive compound, and related compound neoaustin from Penicillium sp. MG-11. Biosci Biotech Bioch. 1994;58:334–8.
Fill TP, Pereira GK, Santos RMG, Rodrigues-Fo E. Four additional meroterpenes produced by Penicillium sp found in association with Melia azedarach. Possible biosynthetic intermediates to austin. Z Naturforsch.2006;62:1035–44.
Arunpanichlert J, Rukachaisirikul V, Phongpaichit S, Supaphon O, Sakayaroj J. Meroterpenoid, isocoumarin, and phenol derivatives from the seagrass-derived fungus Pestaloriopsis sp. PSU-ES194. Tetrahedron. 2015;71:882–8.
Lo HC, et al. Two separate gene clusters encode the biosynthetic pathway for the meroterpenoids austinol and dehydroaustinol in Aspergillus nidulans. J Am Chem Soc. 2012;134:4709–20.
Long Y, et al. Acetylcholinesterase inhibitory meroterpenoid from a mangrove endophytic fungus Aspergillus sp. 16-5c. Molecules. 2017;22:727–34.
Matsuda Y, Awakawa T, Wakimoto T, Abe I. Spiro-ring formation is catalyzed by a multifunctional dioxygenase in austinol biosynthesis. J Am Chem Soc. 2013;135:10962–5.
Fukuyama K, Katsube Y, Ishido H, Yamazaki M, Maebayashi Y. The absolute configuration of desacetylaustin isolated from Emericella nidulans var. dentata. Chem Pharm Bull. 1980;30:2270–1.
Maebayashi Y, Okuyama E, Yamazaki M, Katsube Y. Structure of ED-1 isolated from Emericella dentata. Chem Pharm Bull. 1982;30:1911–2.
Geris R, Rodrigues-Fo E, Garcia da Silva HH, Garcia da Silva I. Larcividal effects of fungal meroterpenoids in the control of Aedes aegypti L., the main vector of dengue and yellow fever. Chem Biodivers. 2008;5:341–5.
Kataoka S, Furutani S, Hirata K, Hayashi H, Matsuda K. Three austin family compounds from Penicillium brasilianum exhibit selective blocking action on cockroach nicotinic acetylcholine receptors. Neurotoxicology. 2011;32:123–9.
Hashimoto N, Zhang WR, Goldstein BJ. Insulin receptor and epidermal growth factor receptor dephosphorylation by three major rat liver protein-tyrosine phosphatases expressed in a recombinant bacterial system. Biochem J. 1992;284:569–76.
Ahmad F, Li PM, Meyerovitch J, Goldstein BJ. Osmotic loading of neutralizing antibodies demonstrates a role for protein-tyrosine phosphatase 1B in negative regulation of the insulin action pathway. J Biol Chem. 1995;270:20504–8.
Cheng A, et al. Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B. Dev Cell. 2002;2:496–503.
Vercauteren M, et al. Improvement of peripheral endothelial dysfunction by protein tyrosine phosphatase inhibitors in heart failure. Circulation. 2006;114:2498–507.
Fang CX, Doser TA, Yang X, Sreejayan N, Ren J. Metallothionein antagonizes aging-induced cardiac contractile dysfunction: role of PTP1B, insulin receptor tyrosine phosphorylation and Akt. Aging Cell. 2006;5:177–85.
Thiebaut PA, Besnier M, Gomez E, Richard V. Role of protein tyrosine phosphatase 1B in cardiovascular diseases. J Mol Cell Cardiol. 2016;101:50–7.
Acknowledgements
This research was supported by a grant from Wonkwang University (2017).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
41429_2018_34_MOESM1_ESM.docx
Furanoaustinol and 7-acetoxydehydroaustinol: New meroterpenoids from a marine-derived fungal strain Penicillium sp. SF-5497
Rights and permissions
About this article

Cite this article
Park, JS., Quang, T.H., Yoon, CS. et al. Furanoaustinol and 7-acetoxydehydroaustinol: new meroterpenoids from a marine-derived fungal strain Penicillium sp. SF-5497. J Antibiot 71, 557–563 (2018). https://doi.org/10.1038/s41429-018-0034-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-018-0034-2
This article is cited by
-
Diversity and Dynamics of Marine Arenicolous Fungi in Three Seasides of the Korean Peninsula
Journal of Microbiology (2023)
-
Quinones from Cordia species from 1972 to 2023: isolation, structural diversity and pharmacological activities
Natural Products and Bioprospecting (2023)
-
Fungal metabolites as anti-diabetic agents: emphasis on PTP1B inhibitors
Phytochemistry Reviews (2021)
-
New preaustinoids from a marine-derived fungal strain Penicillium sp. SF-5497 and their inhibitory effects against PTP1B activity
The Journal of Antibiotics (2019)
