
Abstract
The actinobacterium, strain M26T, was isolated from garden soil that was pre-treated with microwave radiation. The soil sample was collected in Roodepoort, Gauteng Province, South Africa as part of an antibiotic-screening programme. The isolate produced branched vegetative mycelium with sporangiophores bearing small sporangia ranging from 3 to 6 μm in diameter. Rapid genus identification revealed that the isolate belongs to the genus Streptosporangium. To confirm this result, the strain was subjected to polyphasic taxonomic characterisation. Chemotaxonomic characteristics were as follows: meso-DAP in the peptidoglycan, the whole-cell hydrolysate yielded madurose, predominant menaquinones were MK9 (21%), MK9(H2) (40%), MK9(H4) (31%) and MK9(H6) (3%); the polar lipid profile included an aminolipid, phosphoglycolipids, phosphatidylethanolamine, and phosphatidylmonomethylethanolamine. In addition, the fatty acid profile showed the presence of C16:0 (12.8%), C17:1ω8c (14.2%), and 10-methyl-C17:0 (15.8%). Furthermore, 16S rRNA gene sequence phylogenetic analysis showed that the strain is closely related to members of the genus Streptosporangium, which supports its classification within the family Streptosporangiaceae. Strain M26T exhibited antibiosis against a range of pathogenic bacteria, including, but not limited to Acinetobacter baumannii ATCC 19606T, Enterobacter cloacae subsp. cloacae ATCC BAA-1143, Enterococcus faecalis ATCC 51299 (vancomycin resistant), Escherichia coli ATCC 25922, Listeria monocytogenes ATCC 19111, Mycobacterium tuberculosis H37RvT, Pseudomonas aeruginosa ATCC 27853, Salmonella enterica subsp. arizonae ATCC 13314T, and the methicillin-resistant Staphylococcus aureus subsp. aureus ATCC 33591 (MRSA). The name Streptosporangium minutum is proposed with the type strain M26T (=LMG 28850T =NRRL B-65295T).
Similar content being viewed by others


Introduction
The genus Streptosporangium was described by Couch in 1955 [1]. Up until the year 2000, the number of species with validly published names within this genus remained low (10 species). It has only been with the renewed effort in the search for new species that this genus now contains more than 25 described species [2]. Members of this genus are known for their formation of spore vesicles which contain non-motile spores, a fact that separates this genus from other sporangium-producing genera, such as Actinoplanes, Ampullariella, Dactylosporangium, Planomonospora, Planobispora and Spirillospora, all of which have motile spores [3]. The majority of the Streptosporangium species described to date have been isolated from soils, including desert soil [4, 5] and lake sediment [6]. However, several endophytic species have been described, as follows: Streptosporangium oxazolinicum, an endophyte isolated from the root of an orchid [7], Streptosporangium corydalis isolated from the root of Corydalis yanhusuo (a Chinese medicinal herb also known as ‘dark barbarian rope’) [8], and Streptosporangium taraxaci isolated from dandelion root [9].
Members of the genus Streptosporangium are known to produce a range of useful secondary metabolites, which include the antibiotics chloramphenicol, platomycin, and dehydrosinefungin [10]. With the continuous increase in the incidence of antimicrobial-resistant pathogenic microorganisms, there remains a great need for the discovery of novel antimicrobials. In this study, soil samples were collected from different sites in South Africa, actinobacteria isolated using various isolation techniques, and isolates screened for their ability to produce antimicrobial agents. An actinobacterium, which exhibited excellent antimicrobial activity, was isolated from garden soil that had been subjected to a microwave pre-treatment step. To determine the taxonomic position of the isolate, a polyphasic approach was followed.
Materials and methods
Isolation and cultivation
Soil samples were collected from a suburban garden in Roodepoort, Gauteng Province, South Africa and were subjected to a microwave pre-treatment step for the selective isolation of actinobacterial strains [11]. One gram of soil was suspended in 10 ml sterile distilled water and vortexed for 1 min. One millilitre of the suspension was transferred to a sterile glass petri dish, which was placed in the centre of the microwave oven and microwaved for 20 s on the highest power setting (LG model with a 1300 W power output and 950 W microwave output). After the microwave treatment, the soil suspension was allowed to cool, serially diluted in sterile distilled water and spread-plated onto various agar media. All media were supplemented with 100 µg/ml cycloheximide and 100 µg/ml penicillin G. Strain M26T was isolated on Modified Czapek agar (MC agar) [12] after incubation at 30 °C for 21 days. Following isolation, strain M26T was maintained on oatmeal agar (International Streptomyces Project medium 3) supplemented with 0.1% (w/v) yeast extract (ISP3-YE) [13] at room temperature and as a suspension of mycelial fragments in glycerol (20%, v/v) at −20 and −80 °C.
Phenotypic characterisation
The morphological characteristics of strain M26T were determined using standard methods [13]. The isolate was grown on ISP3-YG (ISP3 supplemented with 1.0 g yeast extract, 2.0 g glucose and 2.0 g glycerol per litre) [13] at 30 °C for 14 days, and the morphological characteristics were observed under a light microscope and by cryo-scanning electron microscopy. Standard physiological tests were performed as described previously [13, 14]. For the degradation of Tween 80 and nitrate reduction, the media were supplemented with 0.2% (w/v) yeast extract. ISP media were prepared as described previously [15]. Antibiotic resistance was determined by incorporation of the antibiotics into Bennett’s medium agar plates [16] at the recommended concentrations [14]. Physiological characteristics were determined after growth at 30 °C (unless otherwise stated) for the recommended incubation periods. Carbon source utilisation was initially determined using the recommended C-1 and C-2 media [13], but better results were obtained using ISP medium 9 (ISP9) as the basal medium [15]. All carbon sources for carbon utilisation tests were filter-sterilised and tested at the recommended concentrations [13, 14]. Utilisation of organic acids and resistance to lysozyme were determined as described previously [17] and iodinin production was tested on ISP3-YG agar [13]. For comparative purposes, Streptosporangium canum DSM 45034T, Streptosporangium roseum DSM 43021T, Streptosporangium vulgare DSM 43802T, and Streptosporangium album DSM 43023T were subjected to the same physiological tests. Staining was performed using the standard Gram staining technique.
Chemotaxonomic characterisation
The diaminopimelic acid (DAP) isomer and whole-cell sugar pattern were determined by standard methods [18], with the exception that freeze-dried cells were used instead of colonies from agar plates. Freeze-dried cells were obtained from a 500 ml culture of strain M26T grown in a medium for the enhancement of antibiotic production by Streptosporangium strains (SM, g/L; 10.0 glucose, 10.0 soluble starch, 10.0 glycerol, 2.5 tryptone, 5.0 bacteriological peptone, 2.0 yeast extract, 1.0 NaCl, 3.0 CaCO3, made up to 1 l with tap water, pH 7.3) [19]. The culture was incubated on a rocking shaker at 30 °C for 10 days. Analyses of respiratory quinones, phospholipids and whole-cell fatty acids were carried out by the Identification Service, Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ), Braunschweig, Germany.
Genotypic characterisation
The 16S rRNA gene was amplified by the polymerase chain reaction using the bacterial 16S rRNA gene primers F1 and R5 [20], and the amplified DNA was subjected to a rapid identification method [20]. PCR conditions were as described previously [20]. The amplified DNA was purified for sequencing using a QIAquick PCR purification kit (Qiagen). The sequence was submitted to BLASTn [21] and EzTaxon-e (http://www.ezbiocloud.net) [22] to determine the phylogenetic neighbours of strain M26T. All type strains of the genus Streptosporangium were used for the construction of 16S rRNA gene phylogenetic trees. The software package MEGA (Molecular Evolutionary Genetics Analysis) version 6.01 [23, 24] (http://www.megasoftware.net/) was used. The rooted phylogenetic trees were constructed using the neighbour-joining (NJ) [25], minimum evolution and maximum parsimony (MP) methods [26], and evaluated by bootstrap resampling (1000 replications). Actinomadura madurae DSM 43067T (GenBank accession number: X97889) was used to root the trees. For the gyrB and concatenated gyrB-recA gene analyses, sequence alignments were carried out using MUSCLE [27] in MEGA 6 with default settings. All columns containing gaps were deleted. Phylogenetic trees were constructed using the maximum-likelihood [28], MP [26] and NJ [25] algorithms using the default settings in MEGA 6 for each algorithm. The Kimura two-parameter model [29] was used as the substitution model for the maximum-likelihood and NJ data. Each tree was bootstrapped using 1000 resampled data sets [30]. No filters were applied. GyrB amino acid sequences were determined from gyrB gene sequences translated in silico using DNAMAN version 4.13 (Lynnon BioSoft).
Genome sequencing
Total genomic DNA was isolated by an established method [31] and used in whole genome sequencing. The DNA concentration was determined using a Qubit® Fluorometer (Invitrogen). Genome sequencing was performed by Dr Kirby-McCullough (Department of Biotechnology, University of the Western Cape, Cape Town) on the Illumina Miseq platform, generating 1 595 136 reads. Processing of the Illumina sequencing reads and assembly into a draft genome was performed using the latest version of the A5-miseq assembly pipeline [32]. Quality assessment of the genome was performed using the web-interface of QUAST, developed by the Centre for Algorithmic Biotechnology [33].
Secondary metabolite biosynthetic gene clusters (smBGCs) and possible compounds encoded by the genome sequence were predicted using antibiotics & Secondary Metabolite Analysis SHell (antiSMASH) [34]. Genes were assigned as Clusters of Orthologous Groups (COGs) and sequence similarity detected by BLAST. In addition, comparative genome analysis of strain M26T and its closest phylogenetic neighbours (S. canum CGMCC 4.2126T and S. roseum DSM 43021T) were performed with CGView Comparison Tool (CCT) software (http://stothard.afns.ualberta.ca/downloads/CCT/) [35, 36]. In addition, the genome sequence of strain M26T was submitted as a query genome to the Genome-to-Genome Distance Calculator 2.1 (GGDC; https://ggdc.dsmz.de/ggdc.php). The genome sequence was compared to the reference genomes of S. canum CGMCC 4.2126T and S. roseum DSM 43021T for a genome-based species delineation [37].
DNA–DNA hybridisation
DNA–DNA hybridisation was performed between strain M26T and the type strains of S. album DSM 43023T, S. canum DSM 45034T, S. roseum DSM 43021T, and S. vulgare DSM 43802T. The analysis was performed by the BCCM/LMG culture collection (Belgium). Genomic DNA was isolated using a modification of a published protocol [38]. Hybridisations were performed according to a modification [39, 40] of an established method [41].
Antimicrobial activity assays
Antimicrobial activity was determined by the sloppy-agar overlay technique. For this technique, the isolate was stab-inoculated with sterile toothpicks into SM agar in duplicate. Plates were incubated for 10 days at 30 °C (the duplicate set at 37 °C), and overlaid with 6 ml sloppy agar [42] containing the test bacterium. Strain M26T was also cultured in 500 ml SM for 10 days at 30 °C (160 rpm). The culture was filtered through a coffee filter (House of Coffees, 1 × 4 sized filters). The cell mass of the isolate was sequentially extracted with methanol, chloroform and ethyl acetate, and the culture filtrate sequentially extracted with chloroform and ethyl acetate (Supplementary Figure S1). The 50× concentrated extracts were tested for antimicrobial activity using a modified filter-paper agar disk-diffusion assay [43]. The extracts were spotted onto sterile filter-paper disks (6 mm, Whatman 3 MM, Reeve Angel, NJ, USA) and allowed to dry before being placed onto agar plates inoculated with the test microorganisms. The agar plates were inverted and incubated at each test strain’s optimal growth temperature for 24–36 h and then the diameter of the growth inhibition zones was measured. The antimicrobial assay was performed in duplicate. Negative control filter-paper disks contained extracts from un-inoculated SM medium, while commercial antibiotics (vancomycin and ampicillin; 100 µg/ml) were used as positive controls. In addition, antibacterial activity of the concentrated extracts was also determined by bioautography [44]. The growth conditions of the various test strains are summarised in the supplementary material.
Nucleotide and genome sequence accession numbers
The GenBank accession number for the 16S rRNA gene sequence of Streptosporangium minutum M26T is FJ265773. The accession numbers for the gyrB and recA gene sequences used in this study are listed in Supplementary Table S1. The genome sequence has been deposited at DDBJ/ENA/GenBank under the accession NGFP00000000.
Results and discussion
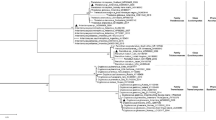
Strain M26T was isolated from garden soil through the use of a selective isolation technique (microwave pre-treatment). A rapid identification technique [20] placed strain M26T within a group containing Streptosporangium species. A 1466 bp 16S rRNA gene sequence was obtained for strain M26T. A search of the EzTaxon-e server showed 99.24% pairwise similarity to S. canum HBUM 170018T, 99.17% to S. vulgare DSM 43802T, 99.17% to S. roseum DSM 43021T and a 99.03% pairwise similarity to S. album DSM 43023T. A phylogenetic tree of Streptosporangium type strains and strain M26T (Fig. 1) showed that strain M26T clustered with the type strains of S. canum, S. album, S. roseum and S. vulgare. This association was supported by a bootstrap value of 85% in the NJ tree and was also seen in the trees constructed using the minimum evolution and MP methods.

16S rRNA gene phylogenetic tree, obtained by the neighbour-joining method, showing the position of strain M26T in the genus Streptosporangium. The 16S rRNA gene sequence of Actinomadura madurae DSM 43067T was used as an out-group. All sequences were edited to the longest common region (1385 bp). GenBank sequence accession numbers are given in parentheses. Numbers at the nodes show the percentage bootstrap values (only values ≥ 50% are shown). Asterisks indicate the clades that were conserved when the neighbour-joining, minimum evolution and maximum parsimony methods were used in constructing the phylogenetic tree. Bar, 5 nt substitutions per 1000 nucleotides
Strain M26T has the IVEAEGR amino-acid signature sequence (S. roseum DSM 43021T GyrB positions 79–85) characteristic of the S. roseum clade in gyrB phylogenetic trees [45]. As expected, phylogenetic analyses based on 1738-nt gyrB gene sequences for 123 members of the family Streptosporangiaceae showed that strain M26T clustered with all of the other Streptosporangium type strains that have the IVEAEGR amino-acid signature sequence: Streptosporangium carneum, Streptosporangium terrae, Streptosporangium jomthongense, Streptosporangium violaceochromogenes, S. album, S. canum, Streptosporangium yunnanense and S. roseum [46]. The association of the nine members of the S. roseum GyrB group was supported by a bootstrap value of 84% in the maximum-likelihood (ML) tree (Supplementary Figure S2). This clustering was also seen in the NJ and MP trees with bootstrap values of 88 and 54%, respectively (data not shown). Strain M26T was very closely associated with the type strains of S. canum, S. yunnanense and S. roseum in all three trees. The bootstrap support for this sub-cluster was very high (100% in all three trees).
The ML phylogenetic tree of the family Streptosporangiaceae based on 123 2632-nt concatenated gyrB-recA gene sequences showed that all 21 type strains of Streptosporangium species with validly published names used in the analysis grouped with very strong bootstrap support (97%; Fig. 2). This association was also seen in the NJ phylogenetic tree (bootstrap value 99%; data not shown), but not the MP phylogenetic tree (data not shown). The genus Planomonospora formed a well-supported group within the evolutionary radiation occupied by the genus Streptosporangium, as has been demonstrated before [47]. Strain M26T clustered with the type strains of S. album, S. roseum, S. canum and S. yunnanense with very strong bootstrap support (100% in the ML tree; Fig. 2). This five-strain cluster was also seen in the NJ and MP trees with very strong bootstrap support (99–100%; data not shown).

Maximum-likelihood phylogenetic tree of the family Streptosporangiaceae based on concatenated gyrB and recA genes. The tree is based on an alignment of 123,2632-nt sequences. Numbers at the nodes show the percentage bootstrap support for each node (only values ≥ 50% are shown). The clades for genera other than Streptosporangium have been compressed (the number in parentheses after each genus name shows the number of sequences used in the analysis of that genus). Asterisks show the nodes that were also obtained in phylogenetic trees constructed with the neighbour-joining and maximum-parsimony algorithms. The scale bar indicates 2 nt substitutions per 100 nucleotides
DNA–DNA hybridisation (DDH) studies showed that strain M26T is a distinct genomic species. DNA re-association values were below the 70% DNA relatedness cutoff recommended [48] for the distinction between prokaryotic genomic species: 40 ± 23% with S. roseum DSM 43021T, 43 ± 6% with S. canum DSM 45034T, 14 ± 4% with S. vulgare DSM 43802T, and 20 ± 14% with S. album DSM 43023T. When the genome sequences were analysed using GGDC 2.1, a DDH estimate of 59.90% [57.1–62.7%] was obtained for strain M26T and S. roseum DSM 43021T, and a DDH estimate of 67.20% [64.2–70%] for strain M26T and S. canum CGMCC 4.2126T. For this reason, a phylogenetic comparison of the genome data of strain M26T with its closest related species was determined (Fig. 3). The genome sequence of S. roseum DSM 43021T was used as a reference genome to build a BLAST atlas to compare the genome sequences of S. canum CGMCC 4.2126T and strain M26T as the genome for S. roseum DSM 43021T is a complete genome, whereas, the genome assemblies for S. canum CGMCC 4.2126T and strain M26T are high quality drafts. Genomic analysis of strain M26T revealed a high degree of similarity between S. canum CGMCC 4.2126T and S. roseum DSM 43021T, in which conserved regions are clearly observed, where 100–90% nucleotide identities are black to light red in colour (Fig. 3a). Coding sequence analysis of strain M26T in comparison to S. canum CGMCC 4.2126T and S. roseum DSM 43021T on the other hand, reveals distinct differences between these strains with a large number of BLAST identities below 50% (Fig. 3b). These results highlight the fact that there are sufficient genetic differences between strain M26T and its phylogenetically closest neighbours S. canum CGMCC 4.2126T and S. roseum DSM 43021T to support its classification as a unique genomic species.

Comparative visualisation of circular genome maps of Streptosporangium roseum DSM 43021T (reference genome) compared with Streptosporangium canum CGMCC 4.2126T and Streptosporangium minutum M26T generated with CGview comparison tool. In the outermost rings, the genes identified by Clusters of Orthologous Groups (COGs; description of COG categories in Supplementary Table S3), followed by blue forward and reverse strand CDSs, tRNAs in orange, rRNA in pink and other RNAs in grey. In the intermediate rings; ring 1 describes the relationship between the reference genome (S. roseum DSM 43021T) and S. canum CGMCC 4.2126 and ring 2 describes the relationship between S. roseum DSM 43021T and strain M26T. a DNA-vs-DNA map: the BLAST hits are coloured according to the percent identities of matches (black to light red, 100–90% identity; blue to light blue, 88–82% identity; and lightest shade of blue, 0% identity). b CDS-vs-CDS map: the BLASTp hits are coloured according to the percent identities of matches (black to light red, 100–50% identity; blue to light blue, 50–10% identity; and lightest shade of blue, 0% identity). BLAST hits are drawn with height proportional to percent identity of the hit. In the innermost rings GC content in black (peaks outside the circle indicate above average and peaks inside the circle indicates below average GC content). GC distributions were measured on the basis of GC skewed using the equation: GC-skew = (G − C) / (G + C), where purple indicates values less than 1 and green indicates values >1. Numbers on the inside denotes the nucleotide positions within the chromosome
Table 1 lists the phenotypic differences between strain M26T and its closest phylogenetic relatives. Growth of the type strain is observed in the presence of cephaloridine (100 μg/ml), oleandomycin (100 μg/ml), penicillin G (10 IU/ml), rifampicin (50 μg/ml) and streptomycin (100 μg/ml), but not in the presence of gentamicin (100 μg/ml), kanamycin (10 μg/ml), lincomycin (100 μg/ml), lysozyme (0.005%, w/v), neomycin (50 μg/ml), tobramycin (50 μg/ml) and vancomycin (50 μg/ml). From cryo-scanning electron microscopy (Fig. 4), the presence of very small sporangia borne on sporangiophores is clearly visible. The sporangiophores were typically 4–6 μm in length and the sporangia had a diameter of 3–6 μm. When plates containing mature sporangia were flooded with water, no spore motility was observed under a light microscope.

Cryo-scanning electron micrograph of strain M26T grown on ISP3-YG agar at 30 °C for 14 days. Sporangiophores bearing sporangia are clearly visible. The bar represents 2 μm
The chemotaxonomic characteristics of strain M26T are consistent with membership of the genus Streptosporangium. Meso-DAP was detected in its peptidoglycan and the whole-cell hydrolysates yielded madurose as the dominant sugar, which is indicative of a cell wall Type III with a Type B sugar pattern (characteristic of the genus Streptosporangium) [49]. The predominant phospholipids detected were phosphatidylethanolamine, phosphatidylmonomethylethanolamine, an aminolipid, three phosphoglycolipids and three unidentified phospholipids (Supplementary Figure S3). MK9 (21%), MK9(H2) (40%), MK9(H4) (31%) and MK9(H6) (3%) were detected as the predominant menaquinones, while C13:0 (1.2%), i-C14:0 (4.2%), C14:0 (1.7%), i-C15:0 (1.1%), C15:0 (4.8%), i-C16:0 (4.8%), C16:1ω9c (2.0%), C16:0 (12.8%), 10-methyl-C16:0 (4.1%), C17:1ω8c (14.2%), C17:1ω6c (2.0%), C17:0 (7.6%), 10-methyl-C17:0 (15.8%), C18:1ω9c (6.1%), C18:0 (2.1%) and 10-methyl-C18:0 (tuberculostearic acid; 3.9%) were the fatty acids detected. These results all support the assignment of strain M26T to the genus Streptosporangium.
Weak to moderate antibiosis was exhibited (in agar overlays) against Burkholderia cepacia ATCC 25416T, Enterobacter cloacae subsp. cloacae ATCC BAA-1143, Escherichia coli ATCC 25922, Mycobacterium aurum A+, Staphylococcus aureus subsp. aureus ATCC 29213, and Salmonella enterica subsp. arizonae ATCC 13314T. Weak bio-activity was detected by filter-paper disk-diffusion assay against Acinetobacter baumannii ATCC 19606T, Bacillus cereus ATCC 10876, E. coli ATCC 25922, Enterococcus faecalis ATCC 29212, E. faecalis ATCC 51299 (vancomycin resistant), Listeria monocytogenes ATCC 19111, methicillin-resistant S. aureus subsp. aureus ATCC 33591, Pseudomonas aeruginosa ATCC 27853, S. enterica subsp. arizonae ATCC 13314T, S. aureus subsp. aureus ATCC 29213, and the yeast test strain Candida krusei ATCC 34135. Very weak bio-activity with regrowth occurring around the filter-paper disk was detected against multidrug-resistant A. baumannii ATCC BAA-1605. Activity against Bacillus subtilis var ING, M. aurum A+, Mycobacterium smegmatis LR222, Mycobacterium tuberculosis H37RvT, and P. aeruginosa ATCC 27853 was observed during bioautography experiments (Supplementary Table S2).
The draft genome size of strain M26T was determined to be 9 581 266 bp (549 scaffolds; N50-score of 30 587 bp) with a G + C mol% of 70.77%. The M26T genome contains 8779 coding sequences (CDSs) of which 8073 are protein-coding genes. Eighty-five RNAs were detected with 21 being rRNA genes, 61 tRNA genes and three non-coding RNA genes. At least three CRISPR arrays were detected in the M26T genome and a total of 706 pseudogenes were detected. Genome sequence analysis of M26T for potential secondary metabolite biosynthetic gene clusters (smBGCs) using the antiSMASH bioinformatics tool led to the identification of 41 BGCs with at least five polyketide synthases (PKSs) of which three were type I PKSs, one type 2 PKS and one type 3 PKS. Twenty non-ribosomal peptide synthetases (NRPSs) were predicted by antiSMASH for M26T, along with four lantipeptides, five terpene clusters, one siderophore cluster, one thiopeptide, one butyrolactone, one bacteriocin, and three gene clusters labelled as “other”. The biosynthetic potential of strain M26T was compared to its phylogenetically closest neighbours for which a genome sequence was available: S. canum CGMCC 4.2126T and S. roseum DSM 43021T (Table 2). It is clear that in comparison to these commonly known natural product producers, strain M26T contains more BGCs, particularly NRPS gene clusters. However, the antiSMASH annotation reveals that at least 39 and 29% of gene clusters from M26T show high similarities to those from the genome of S. canum CGMCC 4.2126T and S. roseum DSM 43021T, respectively. While a high percentage of strain M26T BGCs correspond to known BGCs found in the genomes of S. canum CGMCC 4.2126T and S. roseum DSM 43021T, predictions made with antiSMASH demonstrated that at least 46% of strain M26T secondary metabolite biosynthetic genes encode for new compounds, with ~46% of smBGCs showing low percentage similarity (below 50% homology) to known clusters. Only approximately 7% of smBGCs showed high percentage similarity to known gene clusters. These include a type 3 PKS gene cluster which shares 100% nucleotide sequence similarity with the alkylresorcinol biosynthetic gene cluster from Streptomyces griseus subsp. griseus NBRC 13350; [50,51,52] a type 2 PKS gene cluster, which shares 87% nucleotide sequence similarity to a hexaricin biosynthetic gene cluster from Streptosporangium sp. FXJ7.131; [53] and an NRPS that showed 63% nucleotide sequence similarity to a coelichelin biosynthetic gene cluster from Streptomyces coelicolor A3(2) [54, 55].
On the basis of the polyphasic taxonomic analysis described, strain M26T is proposed to be a novel member of the genus Streptosporangium and is named Streptosporangium minutum (type strain M26T = LMG 28850T = NRRL B-65295T).
Description of Streptosporangium minutum sp. nov
Streptosporangium minutum (mi.nu’tum. L. neut. adj. minutum – very small, referring to the small size of the sporangia).
Gram-staining-positive actinobacterium. Abundant growth is observed on ISP3, ISP3-YE and ISP3-YG. On all three media, red-brown substrate mycelium with light pink aerial mycelium is clearly visible. No growth is observed on ISP2, but sparse growth occurs on inorganic salts-starch agar (ISP4) and on glycerol-asparagine agar (ISP5). A light rose-pink diffusible pigment is produced on ISP3 and a dark red-brown diffusible pigment is produced on ISP3-YG. No iodinin production is observed on ISP3-YG and B vitamins are not required for growth.
Grows in the presence of 0.3% w/v 2-phenylethanol, 0.0001% w/v crystal violet, 3% w/v NaCl (but not 4%) and 0.1% v/v phenol, but not in the presence of sodium azide (0.01% w/v). Weak growth occurs at 4 °C, abundant growth at 30 °C and 37 °C, but no growth at pH 4.3 or at 45 °C. Uses l-histidine, l-hydroxyproline, potassium nitrate, l-serine and l-threonine as sole nitrogen sources, but not DL-α-amino-n-butyric acid, l-arginine, l-cysteine, l-phenylalanine or l-valine. Uses adonitol, d(−) fructose, D(+) galactose, d(+) glucose, inulin, meso-inositol, d(+) lactose, d(+) mannose, d(+) melezitose, d(+) melibiose, raffinose, l(+) rhamnose, ribose, salicin, sucrose, xylitol and d(+) xylose as sole carbon sources. Sodium acetate (0.1%) and sodium citrate (0.1%) are utilised weakly, but L(+) arabinose, d(+) cellobiose, d(−) mannitol and trehalose are not utilised.
H2S production occurs and nitrate is reduced (medium supplemented with 0.2% w/v yeast extract). Lecithinase activity is observed on egg-yolk agar, but not lipase or protease activity. Hippurate is hydrolysed, but pectin is not. Degrades aesculin, arbutin, casein, cellulose, hypoxanthine, starch, Tween 80 (supplemented with 0.2% w/v yeast extract), L-tyrosine and xylan; xanthine is degraded weakly, but adenine, allantoin, gelatin, guanine and urea are not degraded. Utilises the organic acids, sodium acetate, sodium citrate, sodium l-lactate, sodium DL-malate, sodium succinate, and sodium l-tartrate. Sodium benzoate, sodium butyrate, sodium formate, sodium gluconate, sodium maleate, sodium mucate, sodium oxalate, sodium salicylate and sodium sorbate are not utilised.
The whole-cell hydrolysate contains meso-DAP and madurose. The predominant phospholipids are phosphatidylethanolamine, phosphatidylmonomethylethanolamine, an unidentified aminolipid, three unidentified phosphoglycolipids and three unidentified phospholipids. MK9, MK9(H2), MK9(H4) and MK9(H6) are the predominant menaquinones, while C16:0, C17:1ω8c and 10-methyl-C17:0 are the predominant fatty acids. A G + C mol% of 70.77% was determined for the type strain from whole genome sequencing.
The type strain, M26T (=LMG 28850T =NRRL B-65295T), was isolated from soil pre-treated with microwaves.
References
Couch JN. A new genus and family of the Actinomycetales, with a revision of the genus Actinoplanes. J Elisha Mitchell Sci Soc Chapel Hill N C. 1955;71:148–55.
Parte. A List of prokaryotic names with standing in nomenclature. http://www.bacterio.net. Accessed July 2017.
Mertz FP, Yao RC. Streptosporangium carneum sp. nov. isolated from soil. Int J Syst Bacteriol. 1990;40:247–53.
Boubetra D, et al. Streptosporangium algeriense sp. nov., an actinobacterium isolated from desert soil. Int J Syst Evol Microbiol. 2016;66:1034–8.
Chaouch FC, et al. Streptosporangium becharense sp. nov., an actinobacterium isolated from desert soil. Int J Syst Evol Microbiol. 2016;66:2484–90.
Zhang X, et al. Streptosporangium shengliensis sp. nov., a novel actinomycete isolated from a lake sediment. Antonie Van Leeuwenhoek. 2014;105:237–43.
Inahashi Y, Matsumoto A, Õmura S, Takahashi Y. Streptosporangium oxazolinicum sp. nov., a novel endophytic actinomycete producing new antitrypanosomal antibiotics, spoxazomicins. J Antibiot (Tokyo). 2011;64:297–302.
Fang B, et al. Streptosporangium lutulentum sp. nov., Streptosporangium fenghuangense sp. nov. and Streptosporangium corydalis sp. nov., three novel actinobacterial species isolated from National forest park of Fenghuang mountain. Antonie Van Leeuwenhoek. 2016;109:439–48.
Zhao J, et al. Streptosporangium jiaoheense sp. nov. and Streptosporangium taraxaci sp. nov., actinobacteria isolated from soil and dandelion root (Taraxacum mongolicum Hand. _Mazz). Int J Syst Evol Microbiol. 2016;66:2370–6.
Platas G, et al. Nutritional preferences of a group of Streptosporangium soil isolates. J Biosci Bioeng. 1999;88:269–75.
Terekhova L. Isolation of actinomycetes with the use of microwaves and electric. In Kurtböke I, editor. Selective isolation of rare actinomycetes. Queensland: Queensland Complete Printing Services; 2003. pp. 82–101.
Nonomura H, Ohara Y. Distribution of actinomycetes in soil. VIII. Green spore group of Microtetraspora, its preferential isolation and taxonomic characteristics. J Ferment Technol. 1971;49:1–7.
Quintana ET, Goodfellow M. Genus Streptosporangium. In Whitman W, et al., editors. Bergey’s manual of systematic bacteriology: the Actinobacteria. Vol 5. Springer Science and Business Media, New York; 2012. pp. 1811–25.
Locci R. In Williams ST, et al., editors. Bergey’s manual of systematic bacteriology. Baltimore: The Williams & Wilkins Co.; 1989. pp. 2451–508.
Shirling EB, Gottlieb D. Methods for characterisation of Streptomyces species. Int J Syst Bacteriol. 1966;16:313–40.
Atlas RM. Handbook of Microbiological Media. 3rd ed. Boca Raton, FL: CRC Press; 2004.
Gordon RE, Barnett DA, Handerhan JE, Pang C. H-N. Nocardia coeliaca, Nocardia autotrophica, and the nocardin strain. Int J Syst Bacteriol. 1974;24:54–63.
Hasegawa T, Takizawa M, Tanida S. A rapid analysis for chemical grouping of aerobic actinomycetes. J Gen Appl Microbiol. 1983;29:319–22.
Pfefferle C, Theobald U, Gürtler H, Fiedler H-P. Improved secondary metabolite production in the genus Streptosporangium by optimization of the fermentation conditions. J Biotechnol. 2000;80:135–42.
Cook AE, Meyers PR. Rapid identification of filamentous actinomycetes to the genus level using genus-specific 16S rRNA gene restriction fragment patterns. Int J Syst Evol Microbiol. 2003;53:1907–15.
Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402.
Yoon SH, et al. Introducing EzBioCloud: a taxonomically united database of 16S rRNA and whole genome assemblies. Int J Syst Evol Microbiol. 2017;67:1613–7.
Tamura K, et al. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Kumar S, Nei M, Dudley J, Tamura K. MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform. 2008;9:299–306.
Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–25.
Takahashi K, Nei M. Efficiencies of fast algorithms of phylogenetic inference under the criteria of maximum parsimony, minimum evolution, and maximum likelihood when a large number of sequences are used. Mol Biol Evol. 2000;17:1251–8.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–79.
Kimura M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–20.
Felsenstein J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution. 1985;39:783–91.
Marmur J. A procedure for the isolation of deoxyribonucleic acid from microorganisms. J Mol Biol. 1961;3:208–15.
Coil D, Jospin G, Darling AE. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics. 2015;31:587–9.
Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–5.
Blin K, et al. The antiSMASH database, a comprehensive database of microbial secondary metabolite biosynthetic gene clusters. Nucleic Acids Res. 2017;45:D555–D559.
Grant JR, Arantes AS, Stothard P. Comparing thousands of circular genomes using the CGView Comparison Tool. BMC Genom. 2012;13:202.
Stothard P, Grant JR, Van Domselaar G. Visualizing and comparing circular genomes using the CGView family of tools. Brief Bioinform. 2017. https://doi.org/10.1093/bib/bbx081.
Meier-Kolthoff JP, et al. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinforma. 2013;14:60.
Gevers D, Huys G, Swings J. Application of rep-PCR fingerprinting of Lactobacillus species. FEMS Microbiol Lett. 2001;205:31–36.
Goris J, et al. Evaluation of a microplate DNA-DNA hybridization method compared with the initial renaturation method. Can J Microbiol. 1998;44:1148–53.
Cleenwerck I, Vandermeulebroecke K, Janssens D, Swings J. Re-examination of the genus Acetobacter, with descriptions of Acetobacter cerevisiae sp. nov. and Acetobacter malorum sp. nov. Int J Syst Evol Microbiol. 2002;52:1551–8.
Ezaki T, Hashimoto Y, Yabuuchi E. Fluorometric deoxyribonucleic acid-deoxyribonucleic acid hybridization in microdilution wells as an alternative to membrane filter hybridization in which radioisotopes are used to determine genetic relatedness among bacterial strains. Int J Syst Evol Microbiol. 1989;39:224–9.
Sambrook J, et al. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1989.
Eckwall EC, Schottel JL. Isolation and characterization of an antibiotic produced by the scab disease-suppressive Streptomyces diastatochromogenes strain Pon SS II. J Ind Microbiol Biot. 1997;19:2202–25.
Betina V. Bioautography in paper and thin-layer chromatography and its scope in the antibiotic field. J Chromatogr. 1973;78:41–51.
Meyers PR. Gyrase subunit B amino acid signatures for the actinobacterial family Streptosporangiaceae. Syst Appl Microbiol. 2014;37:252–60.
Meyers PR. Molecular-signature analyses support the establishment of the actinobacterial genus Sphaerimonospora (Mingma et al. 2016). Syst Appl Microbiol. 2017;40:423–9.
Meyers PR. Analysis of recombinase A (recA/RecA) in the actinobacterial family Streptosporangiaceae and identification of molecular signatures. Syst Appl Microbiol. 2015;38:567–77.
Wayne LG, et al. International committee on systematic bacteriology. report of the ad hoc committee on reconciliation of approaches to bacterial systematics. Int J Syst Bacteriol. 1987;37:463–4.
Lechevalier MP, Lechevalier H. Chemical composition as a criterion in the classification of aerobic actinomycetes. Int J Syst Bacteriol. 1970;20:435–43.
Ohnishi Y, et al. Genome sequence of the streptomycin-producing microorganism Streptomyces griseus IFO 13350. J Bacteriol. 2008;190:4050–60.
Funabashi M, Funa N, Horinouchi S. Phenolic lipids synthesized by type III polyketide synthase confer penicillin resistance on Streptomyces griseus. J Biol Chem. 2008;283:13983–91.
Hirano S, et al. Conditionally positive effect of the TetR-family transcriptional regulator AtrA on streptomycin production by Streptomyces griseus. Microbiology. 2008;154:905–14.
Tian J, et al. Discovery of pentangular polyphenols hexaricins A–C from marine Streptosporangium sp. CGMCC 4.7309 by genome mining. Appl Microbiol Biot. 2016;100:4189–99.
Bentley SD, et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature. 2002;417:141–7.
Redenbach M, et al. A set of ordered cosmids and a detailed genetic and physical map for the 8 Mb Streptomyces coelicolor A3 (2) chromosome. Mol Microbiol. 1996;21:77–96.
Acknowledgements
Thank you to Di James for DNA sequencing and Miranda Waldron of the Electron Microscope Unit, University of Cape Town (UCT) for help with scanning electron microscopy. Thank you to Hans G. Trüper for assistance with Latin in deriving the specific epithet for strain M26T. The authors also wish to acknowledge the assistance of Dr Kirby-McCullough (University of the Western Cape) in the sequencing of the genome of strain M26T.
Funding:
Marilize le Roes-Hill held a research grant from the National Research Foundation (NRF) of South Africa (grant number: 90304). Paul Meyers was the recipient of research grants from the Medical Research Council of South Africa, and the University Research Committee (UCT). Any opinion, findings and conclusions or recommendations expressed in this material are those of the authors and therefore the NRF does not accept any liability in regard thereto.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest:
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Le Roes-Hill, M., Durrell, K., Prins, A. et al. Streptosporangium minutum sp. nov., isolated from garden soil exposed to microwave radiation. J Antibiot 71, 564–574 (2018). https://doi.org/10.1038/s41429-018-0036-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-018-0036-0
