
Abstract
Three new natural products, including a new anthraquinone derivative isoversicolorin C (1), a new xanthone analog isosecosterigmatocystin (2), and a new amino acid derivative, glulisine A (3), along with six related metabolites (4–9) were isolated from the culture broth and mycelia extracts of the mangrove-derived endophytic fungus Aspergillus nidulans MA-143 under 0.1% ethanol stress. Their structures were elucidated by detailed analysis of their NMR spectra, ECD spectrum, and X-ray crystallographic experiments. Compounds 1 and 4 showed potent antibacterial activity against some of the tested microbes.
Similar content being viewed by others
Introduction
Filamentous fungi are evidenced as a prolific source of secondary metabolites with unique structures and various biological activities. The fungus Aspergillus nidulans is one of the most comprehensively studied and well-characterized species among the genus Aspergillus [1]. Multiple approaches such as cocultivation [2], epigenetic manipulation [3], and genome mining [4, 5], have been developed to activate its cryptic metabolic pathways, and, as a result, a number of new compounds were obtained and reported. During our ongoing study of bioactive secondary metabolites from marine-derived filamentous fungi, the fungal strain A. nidulans MA-143 was obtained as an endophytic fungus from the mangrove plant Rhizophora stylosa. Six new 4-phenyl-3,4-dihydroquinolone derivatives and four new quinazolinone alkaloids were isolated in our previous study from the culture broth and mycelia extracts of this fungus which was cultured in liquid potato–dextrose medium [6, 7]. Considering that A. nidulans MA-143 may have some chemical interactions with the host plant and other endophytic fungi, we carried out a series of experiments trying to activate the silent metabolic genes by adding stress molecules (ethanol and methanol), plant hormones (auxin, abscisic acid, and salicylic acid), and fungal signaling molecules (γ-heptalactones) to the fermentation media. Interestingly, the stressing group (adding 0.1% ethanol) showed more peaks as detected in the HPLC analysis (Fig. 1). We therefore performed a large-scale fermentation of A. nidulans MA-143 in modified minimal (MM) medium under 0.1% ethanol stress, leading to the isolation of a new anthraquinone derivative isoversicolorin C (1), a new xanthone analog isosecosterigmatocystin (2), and a new nitrogen-contained natural product glulisine A (3), together with six known related metabolites, versicolorin C (4) [8], averufin (5) [9], paeciloquinone E (6) [10], averufanin (7) [11], norsolorinic acid (8) [12], and sterigmatocystin (9) [13]. The absolute configuration of compound 1 was determined by TDDFT calculations of its ECD spectra and the chemical structures of compounds 2 and 3 were confirmed by X-ray crystal diffraction analysis. This paper describes the isolation, structure assignment, and biological evaluation of these compounds.

HPLC analysis of extracts from cultures of A. nidulans with (a) or without (b) 0.1% ethanol stress
Results and discussion
Compound 1 was isolated as a yellow amorphous powder. The positive ion ESIMS spectrum showed four peaks at m/z 341.06 [M+H]+, 363.05 [M+Na]+, 698.15 [2M+NH4]+, and 703.10 [2M+Na]+, and its molecular formula was assigned as C18H12O7 by HRESIMS at m/z 341.0648 [M+H]+ (calcd 341.0656), indicating 13 degrees of unsaturation. The UV spectrum of 1 with absorption peaks at 224, 285, 309, and 448 nm represented a typical anthraquinone skeleton [11]. The 1H NMR data of 1 (Table 1) indicated the resonances corresponding to two aliphatic methylenes (H-3′ and H-4′), two aliphatic (H-1′ and H-2′) and three aromatic methines (H-4, H-5, and H-7), and two exchangeable protons (6-OH and 8-OH). The 13C NMR and DEPT spectroscopic data (Table 1) revealed the presence of 18 carbons that were assigned as two methylene carbons (one oxygenated), five methine carbons, and 11 quaternary carbons. Both 1D and 2D NMR data of 1 were similar to that of versicolorin C (4) [8] and 3,6-O-dimethylisoversicolorin C (10) [14]. Compared to compound 4, the chemical shift of C-9 shifted upshield from δC 189.0 in 4 to δC 184.7 in 1, while the resonance of C-4 shifted downshield from δC 101.3 in 4 to 107.2 in 1. These differences suggest that the furan moiety (ring D) in 1 was reverse to that of 4 (Fig. 2). When comparing with that of 3,6-O-dimethylisoversicolorin C (10) [14], signals for two methoxy groups disappeared in that of 1. Additionally, the HMBC correlations from H-1′ to C-1, C-2, C-3′, and C-4′ further confirmed our deduction (Fig. 3). The relative configurations at C-1′ and C-2′ were assigned as cis-orientation based on the coupling constant between H-1′ and H-2′ (3J = 6.3 Hz), which was similar to that of compounds 4 and 9 [8, 13]. The absolute configuration of 1 was determined by electronic circular dichroism (ECD) quantum chemical calculations using Gaussian 09. After geometry optimization of each possible isomers of 1, the minimum energy conformers could be obtained, and the time-dependent density-functional method was used at the B3LYP/6-31 G level to generate calculated ECD spectra of 1. The experimental ECD spectrum of 1 matched well with that calculated for (1′S, 2′R)-1, which showed positive cotton effects (CEs) near 220, 296, and 417 nm and negative CEs around 249, 335, and 391 nm (Fig. 4). The phenyl chromophoric groups might contribute to the CEs near 220 and 249 nm, while the carbonyl chromophoric group contributed to the long-wavelength band around 296 nm. Based on the above discussion, the structure of 1 was assigned and given the trivial name isoversicolorin C.

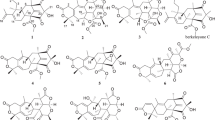
Chemical structures of the isolated compounds 1–9 and the reference compound 10

Key HMBC (arrows) and COSY (bold lines) correlations for compounds 1–3

Measured ECD spectrum of 1 in MeOH compared with the Boltzmann-weighted B3LYP/6-31 G spectra calculated for the solution conformers of (1′S, 2′R)-1 and (1′R, 2′S)-1 in MeOH
Compound 2 was initially obtained as a pale-yellow amorphous powder and had the molecular formula C18H18O8, as determined by HRESIMS at m/z 363.1072 [M+H]+ (calcd 363.1074) and 385.0890 [M+Na]+ (calcd 385.0894), requiring 10 degrees of unsaturation. The 1H NMR data of 2 (Table 1) indicated the presence of one methoxy (H-11), two oxygenated methylenes (H-1′ and H-4′), six methines (including one oxygenated and four aromatic), and one exchangeable proton (5 OH). The 13C NMR and DEPT spectroscopic data of 2 (Table 1) indicated the presence of 18 carbons that were assigned as one methoxy, two oxygenated methylenes, six methines, and nine quaternary carbons. Its planar structure was assigned the same as that of secosterigmatocystin [13] or 3,8-dihydroxy-4-(2,3-dihydroxy-1-hydroxymethylpropyl)-1-methoxyxanthone [15] by discrete analysis of the COSY and HMBC correlations (Fig. 3). However, the 1H and 13C NMR data of 2 were similar but not identical to those of the above-mentioned compounds, which were all measured in DMSO-d6 [13, 15]. Compared to secosterigmatocystin, the chemical shifts of C-2′ and C-3′ shifted from δC 39.8 and 70.8 in secosterigmatocystin to downfield at δC 40.9 and 71.5, respectively, in 2 [13]. But when compared to 3,8-dihydroxy-4-(2,3-dihydroxy-1-hydroxymethylpropyl)-1-methoxyxanthone, the resonances of C-2′ and C-3′ shifted from δC 42.0 and 72.6 in 3,8-dihydroxy-4-(2,3-dihydroxy-1-hydroxymethylpropyl)-1-methoxyxanthone to upfield at δC 40.9 and 71.5, respectively, in 2 [15]. In addition, the optical rotation for 2 was [α] −16.7 (c 0.18, ethanol), while the reported value for secosterigmatocystin was [α]+6.26 (c 0.335, ethanol) [13]. As the configurations of secosterigmatocystin at C-2′ and C-3′ have not been assigned in the previous reports [13, 15], we tried to figure out the stereochemical structure of 2. Upon slow evaporation of 2 in a mixture of methanol and water, quality single crystals were obtained. The relative and absolute configurations of 2 were thus unambiguously established by X-ray diffraction experiment using Cu-Kα radiation (Fig. 5). The refined Flack parameter 0.0(12) allowed for the establishment of the absolute structure of 2 as 2′S and 3′S, and the trivial name isosecosterigmatocystin was assigned to this compound.

X-ray crystallographic structures of compounds 2 and 3
Compound 3 was initially isolated as a white amorphous powder and deduced to possess the molecular formula C11H17N3O2, as determined by HRESIMS at m/z 224.1394 [M+H]+ (calcd 224.1394) and 246.1208 [M+Na]+ (calcd 246.1213), implying five degrees of unsaturation. The 1H NMR data of 3 (Table 1) indicated the presence of two overlapped methyls (H-12 and H-13), three aliphatic methylenes (H-7, H-8, and H-10), and one olefinic methine (H-5). The 13C NMR and DEPT data (Table 1) showed 10 carbon signals corresponding to two overlapped methyls, three methylenes, two methines, and four quaternary carbons. The planar structure of 3 was assigned by analysis of its COSY and HMBC data (Fig. 3), and this compound was derived from the combination of a glutamine and a leucine. Acicular crystals of 3 were obtained in pure methanol and were applied to X-ray diffraction analysis (Fig. 5). The structure of 3 is searchable in the SciFinder database but no literature has been published until now. Herein, we report its spectral and crystal data for the first time and give it the trivial name glulisine A.
Compounds 1–4 were tested for antimicrobial activities against three human pathogens (Escherichia coli, Micrococcus luteus, and Vibrio vulnificus) and four aquatic bacteria (Edwardsiella ictaluri, Vibrio alginolyticus, V. anguillarum, and V. parahaemolyticus). Compound 1 showed potent antibacterial activity against V. alginolyticus and Ed. ictaluri with MIC values of 1 and 4 μg/mL (Table 2), while compound 4 exhibited activity against E. coli, M. luteus, V. alginolyticus, V. parahaemolyticus, and Ed. ictaluri with MIC values ranging from 1 to 8 μg/mL (Table 2), which were comparable to that of the positive control chloramphenicol.
It is well known that the cultivation parameters usually have great influence on the metabolic profile of microorganisms [16]. Various stress conditions have been developed to provocate the cryptic pathways in microorganisms and thus lots of new “cryptic” natural products have been uncovered [17]. In this paper, it is the first time to report that ethanol stress can affect the secondary metabolites of filamentous fungus A. nidulans, leading to the isolation of compounds 1–9 and some of them showed potent antibacterial activities. The precise mechanism that how ethanol stress worked needs to be further studied.
Methods
General experimental procedures
Melting points were determined with an SGW X-4 micro-melting-point apparatus. Optical rotations were measured on an Optical Activity AA-55 polarimeter. UV spectra were measured on a PuXi TU-1810 UV-visible spectrophotometer. ECD spectra were acquired on a Chirascan spectropolarimeter. 1D and 2D NMR spectra were recorded at 500 and 125 MHz for 1H and 13C, respectively, on a Bruker Avance 500 spectrometer with TMS as internal standard. Mass spectra were determined on a VG Autospec 3000 or an API QSTAR Pulsar 1 mass spectrometer. Analytical and semi-preparative HPLC were performed using a Dionex HPLC system equipped with P680 pump, ASI-100 automated sample injector, and UVD340U multiple-wavelength detector controlled by Chromeleon software (version 6.80). Commercially available Si gel (200–300 mesh, Qingdao Haiyang Chemical Co.), Lobar LiChroprep RP-18 (40–63 μm, Merck), and Sephadex LH-20 (Pharmacia) were used for open-column chromatography. All solvents were distilled prior to use.
Fungal material
The endophytic fungus A. nidulans MA-143 (GenBank accession no. JQ839285) was isolated from fresh leaves of the mangrove plant Rhizophora stylosa and was identified by sequence analysis of the ITS region of its rDNA [6]. The strain is preserved at the Key Laboratory of Experimental Marine Biology, Institute of Oceanology, Chinese Academy of Sciences (IOCAS).
Extraction and isolation
Fermentation, extraction, and isolation
For chemical investigations, the fresh mycelia of A. nidulans MA-143 were grown on PDA medium at 28 °C for 6 days and were then inoculated for 30 days at room temperature in 120 × 1-L conical flasks with modified minimal (MM) medium (each flask contained glucose 6.00 g, NaNO3 0.30 g, MgSO4·7H2O 0.30 g, KH2PO4 0.30 g, ZnSO4 0.03 g, ethanol 300 μL, and 300 mL of naturally sourced and filtered seawater, which was obtained from the Huiquan Gulf of the Yellow Sea near the campus of IOCAS, pH 6.5−7.0). The whole-fermented cultures were filtered to separate the broth from the mycelia. The former was extracted with EtOAc three times, while the latter was extracted with a mixture of acetone and H2O (v/v 4:1) three times. The acetone solution was evaporated under reduced pressure to afford an aqueous solution, which was then extracted with EtOAc three times. As the TLC and HPLC profiles of the two EtOAc extracts were nearly identical, they were mixed together and concentrated under reduced pressure to afford an extract (35.7 g). The extract was fractionated by Si gel vacuum liquid chromatography (VLC) using different solvents of increasing polarity from petroleum ether (PE) to MeOH to yield nine fractions (Frs. 1–9) based on TLC and HPLC analysis. Purification of Fr. 4 (4.6 g) by reversed-phase column chromatography (CC) over Lobar LiChroprep RP-18 with a MeOH–H2O gradient (from 10:90 to 90:10) yielded nine subfractions (Frs. 4.1–4.9). Fr. 4.4 (160 mg) was applied to CC on Si gel eluting with a PE–acetone gradient (from 20:1 to 5:1) to yield compound 1 (11.8 mg). Fr. 4.3 (200 mg) was purified by CC on Sephadex LH-20 (MeOH) and then applied to CC on Si gel eluting with a PE–acetone gradient to obtain compound 2 (41.3 mg). Fr. 4.2 (60 mg) was purified by CC on Sephadex LH-20 (MeOH) and then applied to preparative TLC (pTLC) (plate: 20 × 20 cm, developing solvents: CHCl3/MeOH, 20:1) to yield compound 3 (20.1 mg). Fr. 4.5 (1.2 g) was set to CC on Si gel eluting with a PE–acetone gradient (from 25:1 to 2:1) to obtain compounds 4 (17.2 mg) and 9 (38.2 mg). Fr. 4.6 (860 mg) was purified by CC on Si gel eluting with a PE–ethyl acetate (EtOAc) gradient (from 20:1 to 1:1) to obtain compounds 5 (41.5 mg) and 6 (19.7 mg). Fr. 4.8 (920 mg) was purified by CC on Si gel eluting with a PE–EtOAc gradient (from 20:1 to 5:1) to yield compounds 7 (11.6 mg) and 8 (24.3 mg).
Isoversicolorin C (1)
Yellow, amorphous powder; [α] −50.0 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 224 (3.67), 285 (3.45), 309 (2.36), and 448 (1.73); ECD (1.76 mM, MeOH) λmax (Δε) 220 (+0.02), 249 (−0.34), 296 (+0.22), 335 (−0.38), 391 (−0.15), and 417 (+0.02) nm; 1H and 13C NMR data, see Table 1; ESIMS m/z 341.06 [M+H]+, 363.05 [M+Na]+, 698.15 [2M+NH4]+, and 703.10 [2M+Na]+; and HRESIMS m/z 341.0648 [M+H]+ (C18H13O7, calcd 341.0656).
Isosecosterigmatocystin (2)
White, amorphous powder; mp > 350 °C; [α] −16.7 (c 0.18, ethanol); UV (MeOH) λmax (log ε) 231 (3.68), 251 (3.54), and 327 (3.02) nm; ECD (3.31 mM, MeOH) λmax (Δε) 209 (+11.12), 236 (−9.86), 274 (+4.66), 298 (−2.46), 337 (1.02), and 379 (−1.28) nm; 1H and 13C NMR data, see Table 1; HRESIMS m/z 363.1072 [M+H]+ (calcd for C18H19O8, 363.1074); and 385.0890 [M+Na]+ (calcd for C18H18O8Na, 385.0894).
Glulisine A (3)
White, amorphous powder; mp 176–178 °C; UV (MeOH) λmax (log ε) 227 (3.24), 324 (3.46) nm; 1H and 13C NMR data, see Table 1; HRESIMS m/z 224.1394 [M+H]+ (calcd for C11H18N3O2, 224.1394); and 246.1208 [M + Na]+ (calcd for C11H17N3O2Na, 246.1213)
X-ray crystallographic analysis of compounds 2 and 3
All crystallographic data were collected on an Agilent Xcalibur Eos Gemini CCD plate diffractometer equipped with graphite-monochromatic Cu-Kα radiation (λ = 1.54178 Å) at 293(2) K (Crystallographic data of compounds 2 and 3 have been deposited in the Cambridge Crystallographic Data Centre as CCDC 1585394 (for 2) and CCDC 1585396 (for 3). The data can be obtained free of charge via http://www.ccdc.cam.ac.uk/data_request/cif (or from the CCDC, 12 Union Road, Cambridge CB21EZ, UK; fax: +44-1223-336-033; e-mail: deposit@ccdc.cam.ac.uk). The data were corrected for absorption by using the program SADABS [18]. The structures were solved by direct methods with the SHELXTL software package [19]. All non-hydrogen atoms were refined anisotropically. The H atoms were located by geometrical calculations, and their positions and thermal parameters were fixed during the structure refinement. The structures were refined by full-matrix least-squares techniques [19].
Crystal data for compound 2
C18H18O8, F.W. = 362.10, monoclinic space group P2(1), unit cell dimensions a = 28.288(3) Å, b = 7.4028(8) Å, c = 21.1542(19) Å, V = 4252.1(7) Å3, α = 90°, β = 106.285(2)°, γ = 90°, Z = 4, dcalcd = 1.442 mg/m3, crystal dimensions 0.18×0.11×0.05 mm, μ = 1.080 mm–1, and F(000) = 1960. The 7149 measurements yielded 5052 independent reflections after equivalent data were averaged, and Lorentz and polarization corrections were applied. The final refinement gave R1 = 0.1238 and wR2 = 0.3209 [I > 2σ(I)]. The Flack parameter was 0.0(12) in the final refinement for all 7149 reflections with 5052 Friedel pairs.
Crystal data for compound 3
C11H17N3O2, F.W. = 223.28, monoclinic space group P2(1), unit cell dimensions a = 4.9088(4) Å, b = 9.7425(8) Å, c = 12.0775(11) Å, V = 562.90(8) Å3, α = 99.684(2)°, β = 91.5510(10)°, γ = 98.113(2)°, Z = 2, dcalcd = 1.317 mg/m3, crystal dimensions 0.35 × 0.16 × 0.10 mm, μ = 0.093 mm–1, and F(000) = 240. The 2786 measurements yielded 1947 independent reflections after equivalent data were averaged, and Lorentz and polarization corrections were applied. The final refinement gave R1 = 0.0850 and wR2 = 0.1621 [I > 2σ(I)].
Computational methods
Conformational searches for compound 1 were performed via molecular mechanics using the MM + method in HyperChem 8.0 software, and the geometries were further optimized at the B3LYP/6-31 G(d) level via Gaussian 09 software to give the energy-minimized conformers. Then, the optimized conformers were subjected to the calculations of ECD spectra using the TD-DFT at B3LYP/6-31 G (compound 1) level; solvent effects of the MeOH solution were evaluated at the same DFT level using the SCRF/PCM method [20].
Antimicrobial activity assay
Antimicrobial evaluation against three human pathogens (E. coli, M. luteus, and V. vulnificus) and four aquatic bacteria (Ed. ictaluri, V. alginolyticus, V. anguillarum, and V. parahaemolyticus) was carried out by the microplate assay [21]. The human and aquatic pathogens were obtained from the Institute of Oceanology, Chinese Academy of Sciences. Chloramphenicol was used as positive control.
References
Yaegashi J, Oakley BR, Wang CC. Recent advances in genome mining of secondary metabolite biosynthetic gene clusters and the development of heterologous expression systems in Aspergillus nidulans. J Ind Microbiol Biotechnol. 2014;41:433–42.
Schroeckh V, et al. Intimate bacterial-fungal interaction triggers biosynthesis of archetypal polyketides in Aspergillus nidulans. Proc Natl Acad Sci USA. 2009;106:14558–63.
Cichewicz RH. Epigenome manipulation as a pathway to new natural product scaffolds and their congeners. Nat Prod Rep. 2010;27:11–22.
Sanchez JF, et al. Genome-based deletion analysis reveals the prenyl xanthone biosynthesis pathway in Aspergillus nidulans. J Am Chem Soc. 2011;133:4010–7.
Chiang YM, et al. Development of genetic dereplication strains in Aspergillus nidulans results in the discovery of aspercryptin. Angew Chem Int Ed. 2016;55:1662–5.
An CY, et al. 4-phenyl-3,4-dihydroquinolone derivatives from Aspergillus nidulans MA-143, an endophytic fungus isolated from the mangrove plant Rhizophora stylosa. J Nat Prod. 2013;76:1896–1901.
An CY, et al. Aniquinazolines A–D, four new quinazolinone alkaloids from marine-derived endophytic fungus Aspergillus nidulans. Mar Drugs. 2013;11:2682–94.
Hamasaki T, Hatsuda Y, Terashima N, Renbutsu M. The structure of a new metabolite from Aspergillus versicolor. Agr Biol Chem. 1965;29:696–7.
Wu CJ, Li CW, Cui CB. Seven new and two known lipopeptides as well as five known polyketides: the activated production of silent metabolites in a marine-derived fungus by chemical mutagenesis strategy using diethyl sulphate. Mar Drugs. 2014;12:1815–38.
Fredenhagen A, Hug P, Sauter H, Peter HH. Paeciloquinones A, B, C, D, E and F: new potent inhibitors of protein tyrosine kinases produced by Paecilomyces carneus II. Characterization and structure determination. J Antibiot. 1995;48:199–204.
Chen M, Shao CL, Kong CJ, She ZG, Wang CY. A new anthraquinone derivative from a gorgonian-derived fungus Aspergillus sp. Chem Nat Compd. 2014;50:617–20.
Dorairaj S, Banerjee AK, Achari B, Pakrashi SC. Isolation of the anthraquinone norsolorinic acid from Cinchona ledgeriana. Planta Med. 1988;54:469–70.
Zhu F, Lin YC. Three xanthones from a marine-derived mangrove endophytic fungus. Chem Nat Compd. 2007;43:132–5.
Steyn PS, Vleggaar R, Wessels PL, Cole RJ & Scott DB. Structure and carbon-13 nuclear magnetic resonance assignments of versiconal acetate, versiconol acetate, and versiconol, metabolites from cultures of Aspergillus parasiticus treated with dichlorvos. J. Chem. Soc. Perkin Trans. 1979;1:451–9.
Gorst-Allman, CP & Steyn PS Synthesis of model compounds of some biogenetic precursors of aflatoxin B1. J. Chem. Soc. Perkin Trans. 1987;1:163–8.
Scherlach K, Hertweck C. Triggering cryptic natural product biosynthesis in microorganisms. Org Biomol Chem. 2009;7:1753–60.
Pettit RK. Small-molecule elicitation of microbial secondary metabolites. Microb Biotechnol. 2011;4:471–8.
Sheldrick GM. SHELXTL 5.10 for windows NT structure determination software programs. Madison, WI: Bruker Analytical X-Ray System Inc.; 1997.
Sheldrick GM. SHELXL-97 and SHELXS-97 program for X-ray crystal structure solution and refinement. Germany: University of Gottingen; 1997.
Frisch MJ, et al. Gaussian 09, revision D.01. Wallingford, CT: Gaussian, Inc.; 2013.
Pierce CG, et al. A simple and reproducible 96 well plate-based method for the formation of fungal biofilms and its application to antifungal susceptibility testing. Nat Protoc. 2008;3:1494–1500.
Acknowledgements
Financial support from the Natural Science Foundation of China (NSFC grant no. 31570038) and from the NSFC–Shandong Joint Fund for Marine Science Research Centers (U1606403) is gratefully acknowledged. B.-G.W. appreciates the support of Taishan Scholar Project from Shandong Province of China.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
41429_2018_63_MOESM1_ESM.doc
Antibacterial anthraquinone derivatives isolated from a mangrove-derived endophytic fungus Aspergillus nidulans by ethanol stress strategy
Rights and permissions
About this article

Cite this article
Yang, SQ., Li, XM., Xu, GM. et al. Antibacterial anthraquinone derivatives isolated from a mangrove-derived endophytic fungus Aspergillus nidulans by ethanol stress strategy. J Antibiot 71, 778–784 (2018). https://doi.org/10.1038/s41429-018-0063-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-018-0063-x
This article is cited by
-
A new steroid with potent antimicrobial activities and two new polyketides from Penicillium variabile EN-394, a fungus obtained from the marine red alga Rhodomela confervoides
The Journal of Antibiotics (2024)
-
Mangrove-associated endomycota: diversity and functional significance as a source of novel drug leads
Archives of Microbiology (2023)
-
Antimicrobial compounds from marine fungi
Phytochemistry Reviews (2021)
-
Fungal quinones: diversity, producers, and applications of quinones from Aspergillus, Penicillium, Talaromyces, Fusarium, and Arthrinium
Applied Microbiology and Biotechnology (2021)
-
A new acyclic peroxide from Aspergillus nidulans SD-531, a fungus obtained from deep-sea sediment of cold spring in the South China Sea
Journal of Oceanology and Limnology (2020)
