
Abstract
New indanones, designated celludinones A ((±)-1) and B (2), were isolated from the culture broth of the fungal strain Talaromyces cellulolyticus BF-0307. The structures of celludinones were elucidated by spectroscopic data, including 1D and 2D NMR. Celludinone A was found to be a mixture of racemic isomers ((±)-1), which were isolated by a chiral column. Compounds (+)-1 and (−)-1 inhibited the sterol O-acyltransferase (SOAT) 1 and 2 isozymes in a cell-based assay using SOAT1- and SOAT2-expressing Chinese hamster ovary (CHO) cells, while 2 selectively inhibited the SOAT2 isozyme.
Similar content being viewed by others

Introduction
The enzyme sterol O-acyltransferase 2 (SOAT2), predominantly expressed in the liver (hepatocytes) and small intestine [1,2,3,4], is a member of the membrane-bound O-acyltransferase family and regulates cholesterol metabolism in the body [5]. Our recent understanding is that SOAT2 is a more important target for the treatment/prevention of hypercholesterolemia and atherosclerosis than SOAT1 [6,7,8,9]. Although a myriad of SOAT inhibitors have been reported, SOAT2-selective inhibitors are limited [10, 11]. We found that fungal meroterpenoid pyripyropene A (PPPA) is a highly selective inhibitor of SOAT2 [12, 13] and reported that PPPA and several semisynthetic PPPA derivatives were orally active in atherogenic mice [14, 15].
During our continuous screening for SOAT inhibitors of microbial origin using Chinese hamster ovary (CHO) cells expressing African green monkey SOAT1 (SOAT1-CHO cells) and SOAT2 (SOAT2-CHO cells) [12, 13], a culture broth of Talaromyces cellulolyticus BF-0307 was found to selectively inhibit SOAT2 over SOAT1. Three new indanones designated celludinones A1 ((+)-1), A2 ((−)-1), and B (2) were isolated along with known FD-549 (3) [16] from the culture broth (Fig. 1). In the present study, the fermentation, isolation, structural elucidation, and SOAT inhibitory activities of 1–3 are described.

Structures of celludinones and FD-549
Results and discussion
Fungal strain and identification
The fungal strain BF-0307 was isolated from soil collected at Meguro-ku, Tokyo, Japan. In a BLAST search from the International Nucleotide Sequence Databases, BF-0307 had 99.8–100% similarity with the 28S rDNA-D1/D2 and internal transcribed spacer (ITS)-5.8S rDNA of T. cellulolyticus Y-94 (FERMBP5826)T (AB474751) and T. cellulolyticus FERMBP5826T (AB474749) [17, 18]. Therefore, strain BF-0307 was identified with T. cellulolyticus.
Fermentation and isolation of celludinones
Nine-day-old culture broth (2.0 l, pH 6.0) was centrifuged to collect the precipitate. This precipitate was treated with acetone (1.0 l) for 1 h, and acetone extracts were filtered to remove cell debris. The acetone layer was concentrated under reduced pressure to give brown pastes (3.0 g). These pastes were dissolved in a small volume of 40% CH3CN, applied to an ODS column (200 g, i.d. 6 × 20 cm, 100–200 mesh: Fuji Silysia Chemical LTD, Aichi, Japan), and eluted stepwise with 40, 60, 80, and 100% CH3CN (2 fractions per step, 1.0 l each). The 60%-2 and 80%-1 CH3CN fractions containing active materials were evaporated in vacuo to yield crude materials (118 and 191 mg, respectively). The materials from this 60%-2 CH3CN fraction were further purified by HPLC under the following conditions: column, PEGASIL ODS SP100, (i.d. 20 × 250 mm, Senshu Scientific Co., Tokyo, Japan); mobile phase, 40-min linear gradient from 50 to 70% CH3CN; flow rate, 8.0 ml min−1; detection, UV 210 nm. Compound (±)-1 was eluted as a peak with a retention time of 27 min. The fraction of this peak was collected and concentrated in vacuo to yield (±)-1 (6.1 mg) as a pale yellow powder. Materials from the 80%-1 CH3CN fraction were further purified by HPLC: column, PEGASIL ODS SP100 (i.d. 20 × 250 mm), mobile phase, 40-min linear gradient from 65 to 85% CH3CN; flow rate, 8 ml min−1; detection, UV 210 nm. Compounds 2 and 3 were eluted as peaks with retention times of 31 and 18 min, respectively. Each fraction of the peaks was collected and concentrated to yield 2 (7.4 mg) as a red powder and 3 (6.3 mg) as a yellow powder.
Structural elucidation of celludinone A ((±)-1)
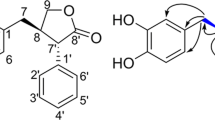
The physicochemical properties of (±)-1 are summarized in Table 1. In the UV spectrum, (±)-1 showed absorption maxima at 261 and 314 nm in MeOH. In the IR spectrum, broad OH absorption at 3414 cm−1, carbonyl absorption at 1697 cm−1, and aromatic C–C stretch absorption (for carbon–carbon bonds in the benzene ring) at 1617 cm−1 were observed. The molecular formula C23H26O5 was assigned based on HR-ESIMS (m/z, found 383.1850, calcd 383.1858 for C23H27O5 [M + H]+), indicating 11 degrees of unsaturation. The 13C NMR spectrum of (±)-1 in CDCl3 (Table 2) showed 23 resolved signals, which were classified into five methyl carbons, one methylene carbon, two sp [3] quaternary carbons, 5 sp2 methine carbons, 9 sp2 quaternary carbons, and one carbonyl carbon by an analysis of heteronuclear multiple-quantum correlation (HMQC) data (Table 2). The 1H-NMR spectrum of (±)-1 in CDCl3 showed 22 proton signals (Table 2). The connectivity of proton and carbon atoms was established by HMQC. As shown in Fig. 2, analyses of the 1H–1H COSY spectrum and 13C–1H long-range couplings of 2J and 3J observed in the HMBC spectrum gave the following two partial structures: I and II. The cross peaks from 22-H3 (δ 0.99) to C-1 (δ 56.0), C-2 (δ 208.0), C-9 (δ 84.4), and C-23 (δ 22.2), from 23-H3 (δ 1.35) to C-1, C-2, C-9, and C-22 (δ 21.5), from 4-H (δ 7.16) to C-2, C-6 (δ 122.8), C-8 (δ 137.0), and C-21 (δ 21.4), from 6-H (δ 6.90) to C-4 (δ 116.2), C-8, and C-21, and from 21-H3 (δ 2.36) to C-4, C-5 (δ 142.1), and C-6 supported the presence of partial structure I. The cross peaks from 13-H (δ 6.89) to C-11 (δ 153.9), C-15 (δ 151.9), and C-16 (δ 28.8) and from 14-H (δ 6.11) to C-10 (δ 111.8), C-12 (δ 121.8), and C-15 supported the presence of 1,2,3,4-tetrasubstituted benzene ring A. Furthermore, the cross peaks from 16-H2 (δ 3.30) to C-11, C-12, C-13 (δ 129.8), and C-18 (δ 133.9), from 17-H (δ 5.33) to C-19 (δ 18.0) and C-20 (δ 25.9), from 19-H3 (δ 1.76) to C-17 (δ 122.5) C-18, and C-20, (δ 25.9) and from 20-H3 (δ 1.79) to C-17, C-18, and C-19 supported the presence of an isoprenyl moiety. The cross peaks from 16-H2 to C-12 indicated that C-12 of ring A was bound to C-16 of the isoprenyl moiety, forming partial structure II. By taking the molecular formula, IR data, and chemical shifts in C-7 (δ 155.1), C-9, C-11, and C-15 into consideration, a hydroxyl moiety should be bound to these four carbons. Although there was no direct correlation between partial structures I and II, these structures including the linkage of C-8–C-9–C-10 appeared to be connected from the molecular formula, degrees of unsaturation, and chemical shifts of C-8, C-9, and C-10. Taken together, the structure of (±)-1 was elucidated as shown in Fig. 1.

Key correlations in 1H–1H COSY and HMBC spectra of (±)-1
Separation of (+)- and (−)-celludinone A (1)
Although 1 has a chiral carbon at C-9, the CD spectrum of 1 was almost flat (Fig. 3a). This result suggested that 1 was a racemic mixture. Therefore, 1 (2.8 mg) was further purified by HPLC using a chiral column. As a result, (+)-1 and (−)-1 were eluted as peaks with retention times of 11 min and 12 min, respectively (Fig. 4). These peaks were collected and concentrated to yield (+)-1 (1.1 mg) as an orange powder and (−)-1 (1.2 mg) as a yellow powder.

CD spectra of celludinones A ((±)-1), A1 ((+)-1), and A2 ((−)-1). a Celludinones A ((±)-1), b A1 ((+)-1), and c A2 ((−)-1)

Separation of (±)-1 with a chiral column. Celludinone A ((±)-1) was subjected to HPLC with a chiral column to separate celludinones A1 ((+)-1) and A2 ((−)-1)
Absolute stereochemistries of (+)-1 and (−)-1
In order to elucidate absolute configurations, the CD spectra of (+)-1 and (−)-1 were measured. As shown in Fig. 3b, the CD curve of (+)-1 exhibited the first negative Cotton effect at 230 nm and the second positive Cotton effect at 215 nm, which was similar to that of (S)-1-mesityl-2,2-dimethyl-2,3-dihydro-1H-indeno-1-ol (the first negative Cotton effect at 210 nm and the second positive Cotton effect at 200 nm) [19]. Conversely, the CD curve of (−)-1 exhibited the first positive Cotton effect at 230 nm and the second negative Cotton effect at 215 nm (Fig. 3c), which was similar to that of (R)-1-(3-methoxy-2,4,6-trimethylphenyl)-2,2-dimethyl-2,3-dihydro-1H-inden-1-ol (the first negative Cotton effect at 200 nm and the second positive Cotton effect at 210 nm) [19]. Accordingly, the absolute configurations of the hydroxyl moiety in (+)-1 and (−)-1 were elucidated to be 9S and 9R, respectively.
Structural elucidation of 2
The physicochemical properties of 2 are summarized in Table 1. In UV spectra, 2 showed an absorption maximum at 264 nm in MeOH. In IR spectra, broad OH absorption at 3414 cm−1, typical C–H (CH2) stretching absorption at 2923 cm−1, carbonyl absorption at 1697 cm−1, and aromatic C–C stretch absorption (for carbon–carbon bonds in the benzene ring) at 1617 cm−1 were observed. The molecular formula C40H38O8 was assigned based on HR-ESIMS (m/z, found 647.2646, calcd. 647.2644 for C40H39O8 [M + H]+), indicating 22 degrees of unsaturation. The 13C NMR spectrum of 2 in DMSO-d6 showed 40 resolved signals, which were classified into 6 methyl carbons, 2 methylene carbons, 10 sp2 methine carbons, 20 sp2 quaternary carbons, and 2 carbonyl carbons by an analysis of HMQC data (Table 3). The 1H-NMR spectrum of 2 in DMSO-d6 showed 37 proton signals (Table 3). The connectivity of protons and carbon atoms was established by HMQC. As shown in Fig. 5, analyses of the 1H–1H COSY spectrum and 13C–1H long-range couplings of 2J and 3J observed in the HMBC spectrum gave the following two partial structures: III and IV. The cross peaks from 14-H2 (δ 3.12) to C-16 (δ 131.5), from 15-H (δ 5.24) to C-17 (δ 17.7) and C-18 (δ 25.6), from 17-H3 (δ 1.65) to C-15 (δ 25.9), C-16 (δ 131.5), and C-18, and from 18-H3 (δ 1.67) to C-15, C-16, and C-17 supported the presence of an isoprenyl moiety. Furthermore, cross peaks from 5-H (δ 7.09) to C-3 (δ 159.4) and C-7 (δ 159.4) and from 6-H (δ 6.24) to C-2 (δ 113.0), C-4 (δ 118.0), and C-7 supported the presence of 1,2,3,4-tetrasubtituted benzene ring B. The following linkages from 5-H to C-14 (δ 27.0) and from 14-H2 to C-3, C-4, and C-5 (δ 136.2) proved the correlation of ring B and the isoprenyl moiety. Cross peaks from 10-H (δ 6.47) to C-8 (δ 127.0), C-9 (δ 155.0), C-12 (δ 121.3), and C-19 (δ 21.0), from 12-H (δ 6.24) to C-8 and C-10 (δ 115.7) and from 19-H3 (δ 21.0) to C-10, C-11 (δ 140.3), and C-12 supported the presence of 1-methyl,3,4,5-trisubtituted benzene ring C. Since the long-range couplings of 4J were observed from 6-H and 10-H to the carbonyl carbon C-1 (δ 200.5), the two substructures should be connected via C-1 as shown in Fig. 5 (partial structure III) [16]. The remaining signals of partial structure IV were very similar to those of 1, except for two missing methyl signals (22-H3 and 23-H3). Furthermore, the molecular formula, degrees of unsaturation, and chemical shifts indicated that 2 had six hydroxyl moieties. In order to confirm the number of hydroxyl groups, 2 was methylated with trimethylsilyldiazomethane [20]. Reaction products were analyzed by LC-MS. As a result, tri-methylated 2 to hexa-methylated 2 were observed (Supplemental Fig. 1). Of these, hexa-methylated 2 was eluted as a peak with a retention time of 8.97 min ([M + H]+ 731). Thus, the structure of celludinone B (2) was elucidated as shown in Fig. 1, and fulfilled the molecular formula and degrees of unsaturation.

Key correlations in 1H–1H COSY and HMBC spectra of 2
Structural identification of 3
Based on spectral data, including 1H-NMR, 13C NMR, and MS, and the search results of SciFinder Scholar, 3 was identified as the known benzophenone FD-549 [16] (Fig. 1).
Inhibition of SOAT isozymes using SOAT1- and SOAT2-CHO cells
The effects of 1–3 on SOAT1 and SOAT2 isozymes (African green monkey) were evaluated in a cell-based assay using SOAT1- and SOAT2-CHO cells [12, 13, 21]. As shown in Table 4, (±)-1 inhibited the SOAT1 and SOAT2 isozymes with IC50 values of 12 and 9.9 µM, respectively, giving an SI value of 0.084 ranging between −1.0 and +1.0 (dual-type inhibition). After the separation of (±)-1 by chiral HPLC, (+)-1 and (−)-1 exhibited similar SOAT inhibitory activities and selectivities to (±)-1, suggesting that the 9-OH configuration of 1 does not affect SOAT inhibitory activity. On the other hand, 2 including benzophenone moieties showed SOAT2-selective inhibition; respective IC50 values for SOAT1 and SOAT2 were 2.8 and 0.15 µM, giving an SI value of +1.27 (>+1.0 means SOAT2-selective inhibition). The respective IC50 values of 3 were 9.9 and 0.91 µM, giving an SI value of 1.0. Thus, 2 and 3 were both SOAT2-selective inhibitors. These findings strongly suggested that the common benzophenone moieties in 2 and 3 were important for SOAT2-selective inhibition. Very similar results to SOAT inhibition were observed when human SOAT1- and SOAT2-expressing CHO cells were used for assays (Table 4). Compounds 1 to 3 did not exert cytotoxic effects in these cell lines, even at 20 µM. So far, we have discovered a lot of SOAT isozyme inhibitors showing huge structural diversity from microorganisms such as pyripyropenes and beauveriolides [10, 22]. Compounds 1 and 2 are the first indanones possessing SOAT inhibitory activity.
Materials and methods
General
Various NMR spectra were obtained using the NMR System 400 MHz spectrometer (Agilent Technologies, Santa Clara, CA, USA). FAB-MS spectra were recorded on a mass spectrometer (JMS-700 Mstation; JEOL, Tokyo, Japan). Optical rotations were measured with a digital polarimeter (DIP-1000; JASCO, Tokyo, Japan). UV spectra were recorded on a spectrophotometer (8453 UV-visible spectrophotometer; Agilent Technologies). IR spectra were recorded on a Fourier transform IR spectrometer (FT-710; Horiba Ltd., Kyoto, Japan). CD spectra were recorded with a CD spectrometer (J-720 spectropolarimeter, JASCO, Tokyo, Japan). An LC-MS analysis was performed using the AccuTOF LC-plus JMS-T100LP system (JEOL, Tokyo, Japan).
Materials
[1-14C]Oleic acid (1.85 GBq mmol−1) was purchased from PerkinElmer (Waltham, MA, USA). Fetal bovine serum was purchased from Biowest (Nuaille, France). Dulbecco’s modified Eagle’s medium and Hank’s buffered salt solution were purchased from Nissui Pharmaceutical Co. (Tokyo, Japan). GIT medium was from Nippon Seiyaku Co. (Tokyo, Japan). Penicillin (10,000 units ml−1), streptomycin (10,000 mg ml−1), and glutamine (200 mm) solution were from Invitrogen (Carlsbad, CA, USA). Phosphatidylcholine, phosphatidylserine, dicetylphosphate, cholesterol, Ham’s F-12 medium, malachite green oxalate, and BFA1 were purchased from Sigma-Aldrich (St Louis, MO, USA). Perchloric acid and Triton X-100 were purchased from Wako (Osaka, Japan).
Fungal strain and identification
Fungal strain BF-0307 was isolated from soil collected from Meguro-ku, Tokyo, Japan, and was identified as T. cellulolyticus by TechnoSuruga Laboratory Co., Ltd. (Shizuoka, Japan).
Fermentation of T. cellulolyticus BF-0307
A loopful of spores of strain BF-0307 was inoculated into 100 ml seed medium consisting of 2.4% potato dextrose broth (Becton Dickinson and Company, Franklin Lakes, NJ) and 0.1% agar (adjusted to pH 6.0 before sterilization) on a 500-ml Erlenmeyer flask. The inoculated Erlenmeyer flask was incubated on a rotary shaker (180 rpm) at 27 °C for 4 days in order to obtain the seed culture. In the production of (±)-1 to 3, the culture was initiated by transferring 1 ml of the seed culture into 20 500-ml Erlenmeyer flasks containing 100 ml production medium (1.0% galactose, 2.0% glycerol, 1.0% glucose, 0.25% tryptone, 0.25% yeast extract, 0.05% KH2PO4, and 0.01% FeSO4•7H2O adjusted to pH 6.0 before sterilization). Fermentation was performed on a rotary shaker (180 rpm) at 27 °C for 9 days.
Separation of (+)- and (−)-celludinone A (1)
Celludinone A (1) (2.8 mg) was separated by HPLC using a chiral column under the following conditions: column, CHIRALPAK IA, (i.d. 4.6 × 150 mm, DAICEL Corporation, Tokyo, Japan); mobile phase, isocratic conditions of 46% CH3CN; flow rate, 1.0 ml min−1; detection, UV260 nm.
Methylation of celludinone B (2)
Celludinone B (2) (1.8 mg) dissolved in MeOH (500 µl) and trimethylsilyldiazomethane (750 µl, Tokyo Industry Co., Ltd., Tokyo, Japan) were mixed at 25 °C. After 30 min, the reaction products were analyzed by LC-MS under the following conditions: column, CAPCELLCORE C-18, (i.d. 2.1 × 50 mm, OSAKA SODA, Osaka, Japan); mobile phase, 8-min linear gradient from 50 to 100% CH3CN-0.1%HCOOH and 7 min of 100% CH3CN-0.1%HCOOH; flow rate, 0.4 ml min−1; detection, UV 210 nm.
Cell culture
CHO cells (AC29 cells, SOAT-deficient cells) expressing the SOAT1 or SOAT2 gene from the African green monkey and humans were cultured by a previously described method [12].
Assay for SOAT activity in SOAT1- and SOAT2-CHO cells
Assays for SOAT1 and SOAT2 activities using SOAT1- and SOAT2-CHO cells were performed using our established method [13, 21]. Briefly, SOAT1- or SOAT2-CHO cells (1.25 × 105 cells in 250 µl of medium) were cultured in a 48-well plastic microplate in the culture medium described above and allowed to recover at 37 °C overnight in 5% CO2. Assays were performed with cells that were at least 80% confluent. Following the overnight recovery, a test sample (2.5 µl MeOH solution) and [1-14C]oleic acid (5 µl 10% EtOH/PBS solution, 1 nmol, 1.85 KBq) were added to each culture. After a 6-h incubation at 37 °C in 5% CO2, medium was removed, and the cells in each well were washed twice with PBS. Cells were lysed by adding 0.25 ml of 10 mM Tris-HCl (pH 7.5) containing 0.1% (w/v) sodium dodecyl sulfate, and [14C]CE was analyzed with a FLA-7000 analyzer (Fuji Film). In this cell-based assay, [14C]CE was produced by the reaction of SOAT1 or SOAT2. SOAT inhibitory activity (%) is defined as ([1-14C]CE-drug/[14C]CE-control) × 100. The IC50 value is defined as the drug concentration causing the 50% inhibition of biological activity.
References
Alger HM, et al. Inhibition of acyl-coenzyme A:cholesterol acyltransferase 2 (ACAT2) prevents dietary cholesterol-associated steatosis by enhancing hepatic triglyceride mobilization. J Biol Chem. 2010;285:14267–74.
Oelkers P, Behari A, Cromley D, Billheimer JT, Sturley SL. Characterization of two human genes encoding acyl coenzyme A:cholesterol acyltransferase-related enzymes. J Biol Chem. 1998;273:26765–71.
Cases S, et al. ACAT-2, a second mammalian acyl-CoA:cholesterol acyltransferase. Its cloning, expression, and characterization. J Biol Chem. 1998;273:26755–64.
Parini P, et al. ACAT2 is localized to hepatocytes and is the major cholesterol-esterifying enzyme in human liver. Circulation. 2004;110:2017–23.
Rudel LL, Lee RG, Cockman TL. Acyl coenzyme A:cholesterol acyltransferase types 1 and 2: structure and function in atherosclerosis. Curr Opin Lipidol. 2001;12:121–7.
Buhman KK, et al. Resistance to diet-induced hypercholesterolemia and gallstone formation in ACAT2-deficient mice. Nat Med. 2000;6:1341–7.
Willner EL, et al. Deficiency of acyl CoA:cholesterol acyltransferase 2 prevents atherosclerosis in apolipoprotein E-deficient mice. Proc Natl Acad Sci USA. 2003;100:1262–7.
Yagyu H, et al. Absence of ACAT-1 attenuates atherosclerosis but causes dry eye and cutaneous xanthomatosis in mice with congenital hyperlipidemia. J Biol Chem. 2000;275:21324–30.
Fazio S, et al. Increased atherosclerosis in LDL receptor-null mice lacking ACAT1 in macrophages. J Clin Invest. 2001;107:163–71.
Ohshiro T, Tomoda H. Isoform-specific inhibitors of ACATs: recent advances and promising developments. Future Med Chem. 2011;3:2039–61.
Ohshiro T, Tomoda H. Acyltransferase inhibitors: a patent review (2010-present). Expert Opin Ther Pat. 2015;25:145–58.
Lada AT, et al. Identification of ACAT1- and ACAT2-specific inhibitors using a novel, cell-based fluorescence assay: individual ACAT uniqueness. J Lipid Res. 2004;45:378–86.
Ohshiro T, Rudel LL, Omura S, Tomoda H. Selectivity of microbial acyl-CoA: cholesterol acyltransferase inhibitors toward isozymes. J Antibiot. 2007;60:43–51.
Ohshiro T, et al. Pyripyropene A, an acyl-coenzyme A:cholesterol acyltransferase 2-selective inhibitor, attenuates hypercholesterolemia and atherosclerosis in murine models of hyperlipidemia. Arterioscler Thromb Vasc Biol. 2011;31:1108–15.
Ohshiro T, et al. New pyripyropene A derivatives, highly SOAT2-selective inhibitors, improve hypercholesterolemia and atherosclerosis in atherogenic mouse models. J Pharmacol Exp Ther. 2015;355:299–307.
Kyo T, Kawamura Y, Okazaki T, Mizogami K, Morimoto S. A benzophenone compound FD-549. Japan: Taisho Pharma Co Ltd; 1996.
Fujii T, Hoshino T, Inoue H, Yano S. Taxonomic revision of the cellulose-degrading fungus Acremonium cellulolyticus nomen nudum to Talaromyces based on phylogenetic analysis. FEMS Microbiol Lett. 2014;351:32–41.
Fujii T, Koike H, Sawayama S, Yano S, Inoue H. Draft genome sequence of talaromyces cellulolyticus strain Y-94, a source of lignocellulosic biomass-degrading enzymes. Genome Announc. 2015;3:e00014–00015.
Casarini D, Mancinelli M, Mazzanti A, Boschi F. Stereodynamics and absolute configuration of stereolabile atropisomers in 2,2-dimethyl-1-aryl-1-indanols. Chirality. 2011;23:768–78.
Aoyama T, Shioiri T. New methods and reagents in organic synthesis. 8. Trimethylsilyldiazomethane. A new, stable, and safe reagent for the classical arndt-eistert synthesis. Tetrahedron Lett. 1980;21:4461–2.
Ohshiro T, et al. Selective inhibition of sterolO-acyltransferase 1 isozyme by beauveriolide III in intact cells. Sci Rep. 2017;7:4163.
Tomoda H, Omura S. Potential therapeutics for obesity and atherosclerosis: inhibitors of neutral lipid metabolism from microorganisms. Pharmacol Ther. 2007;115:375–89.
Acknowledgements
We wish to thank Ms. Noriko Sato and Dr. Kenichiro Nagai (Graduate School of Pharmaceutical Sciences, Kitasato University) for the measurements of NMR spectra and MS data, and Prof. L.L. Rudel (Wake Forest University, Winston-Salem, NC, USA) for kindly providing SOAT1-CHO and SOAT2-CHO cells. This work was supported by JSPS KAKENHI Grant numbers JP26253009 (HT) and JP16K18900 (TO), the Takeda Science Foundation (HT), and a Kitasato University Research Grant for Young Researchers (TO).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Ohshiro, T., Seki, R., Fukuda, T. et al. Celludinones, new inhibitors of sterol O-acyltransferase, produced by Talaromyces cellulolyticus BF-0307. J Antibiot 71, 1000–1007 (2018). https://doi.org/10.1038/s41429-018-0097-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-018-0097-0
This article is cited by
-
New dihydronaphthothiophene derivatives by the biological transformation of seriniquinone using marine-derived actinomycete Streptomyces albogriseolus OM27-12
The Journal of Antibiotics (2022)
-
Voluhemins, new inhibitors of sterol O-acyltransferase, produced by Volutella citrinella BF-0440
The Journal of Antibiotics (2020)
