
Abstract
In recent years, incidences of invasive fungal infections have greatly increased, especially in immunosuppressed patients, but most today's antifungal drugs are not completely effective due to the development of drug resistance, as well as potential toxicity and adverse effects. Consequently, it is imperative to search for novel antifungal agents to combat fungal infections. This review will discuss the advances in the traditional antifungal therapy, and present an overview of novel strategies for the treatment of fungal infections. The papers presented here highlight new targets that could be exploited for development of new antifungal agents.
Similar content being viewed by others

Introduction
Fungal infections are responsible for over one million human deaths per annum, which is a significant worldwide health problem [1]. In recent decades, the number of opportunistic fungal infections had greatly increased in immunosuppressed patients and those admitted in intensive care units [2,3,4]. These infections range from non-life-threatening mucocutaneous illnesses to invasive infections involved in virtually any organ [5]. The fungal genera most often associated with the invasive fungal infections, including species of Aspergillus, Candida, and Cryptococcus [5, 6]. Of the Candida species isolated from humans, Candida albicans is the most commonly encountered human fungal pathogen [7]. Systemic candidiasis, caused by C. albicans, had become one of the main causes of death in deep fungal infection patients [3]. However, infections caused by Aspergillus fumigatus and Cryptococcus neoformans were also common [8].
Only few classes of antifungals, such as polyenes, azoles, echinocandins, allylamines, and flucytosine were available for the treatment of fungal infections [8, 9]. Unfortunately, these antifungals had various drawbacks in terms of toxicity, spectrum of activity, safety, and pharmacokinetic properties [10]. Besides, with the long-term and large-scale application of these antifungal agents, there had been a notable increase in drug resistance [3]. Consequently, it was critical to search for new antifungal drugs, and devise innovative strategies to combat fungal infections.
The papers presented here the advances in the traditional antifungal therapy and highlighted new targets that could be exploited for development of new antifungal agents. These new targets, which included homoserine transacetylase, methionine synthase, ATP sulfurylase, transcription factor protein (MET4), homocysteine synthase, aspartate kinase, homoserine dehydrogenase, homoserine kinase, threonine synthase, acetolactate synthase, ROS production, biofilm formation, sulfite transporter, phosphopantetheinyl transferase, mitochondrial phosphate carrier, and bromodomain (BD), show great promise as antifungal targets.
Traditional antifungal agents
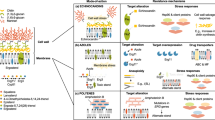
Currently available antifungal agents are mainly divided into six categories: azoles, allylamines/thiocarbamates, morpholines, polyenes, pyrimidine analogs, and echinocandins [10]. These antifungal agents usually targeted ergosterol biosynthesis pathway, fungal cell wall, or fungal DNA/RNA.
Azoles are the most widely used class of antifungal drugs (Fig. 1) [11, 12]. Azoles exerted antifungal effect by inhibition of cytochrome P450-dependent 14α-lanosterol demethylase (Cyp51) encoded by the ERG11 gene that converted lanosterol to ergosterol [10, 13]. Moreover, fluconazole (FLC, 1) and itraconazole (2) also resulted in the accumulation of obtusifolione in the ergosterol biosynthetic pathway, mainly due to the inhibition of NADPH-dependent 3-ketosteroid reductase (ERG 27), which catalyzed the reduction of the 3-ketosteroid obtusifolione to obtusifoliol in C. neoformans [11, 14]. The increased use of azoles had led to an increase in drug resistance, limiting their effectiveness [15]. However, the development of new azoles agents was still an active area in current antifungal drug research. Isavuconazole (4) was approved in 2015 for treatment of invasive aspergillosis and mucormycosis [16]. Isavuconazole possessed a broad range of antifungal activity and good water solubility and displayed improved safety and tolerability comparing with voriconazole [17]. Another new azoles albaconazole (3) under clinical evaluations, showed potent activities against clinically resistant fungal strains [16].

Structures of antifungal reagents target ergosterol biosynthesis pathway
Allylamines (e.g., terbinafine, 5) and thiocarbamates (e.g., tolnaftate, 6) inhibited the squalene epoxidase encoded by the ERG1 gene that converted squalene to 2,3-squalene epoxide [10, 14]. Morpholines (e.g., amorolfine, 7) exerted antifungal effect by inhibition Δ8-Δ7 isomerase (ERG2) and the Δ14-reductase (ERG24) involved in ergosterol biosynthesis [10, 18]. However, all of these drugs have been mostly used for the control of dermatophyte fungal infections [10, 14, 18].
Polyenes (Fig. 2) are actinomycetes-derived unsaturated macrolactones that complex with ergosterol and disrupt the fungal plasma membrane, which result in increase of membrane permeability, the leakage of vital cytoplasmic components, and ultimately death of the fungal cell [14, 19]. Amphotericin B (AMB, 8), natamycin (9), and nystatin (10) were the only three polyenes in clinical use [10]. However, one of the primary drawbacks of polyenes was their significant toxicity [12]. Although nephrotoxicity could be reduced with the use of lipid formulations of AMB, it still occurred, especially with higher doses or prolonged administration [13, 20]. Subsequently, researchers discovered the novel polyene compound SPK-843 (11). SPK-843 showed less renal toxicity than both AMB or liposomal AMB and also better activity than micafungin in vivo [21]. Unfortunately, SPK-843 had not been in clinical trials for many years (phase III clinical study, efficacy, safety, and pharmacokinetics of SPK-843 in the treatment of cryptococcosis or aspergillosis infections in 2011: Drugbank).

Structures of polyenes antifungal reagents
Pyrimidine analogs, 5-fluorocytosine (5-FC) and 5-fluorouracil (5-FU), were the synthetic structural analogs of nucleotide cytosine [14]. 5-FC worked as an antifungal agent through conversion to 5-FU within target cells. 5-FU incorporated into RNA resulting in disruption of protein synthesis, and also inhibited DNA synthesis and the nuclear division through effect on thymidylate synthase [10, 18, 19]. 5-FC exhibited activity against some strains of Candida and Cryptococcus. However, the most filamentous fungi lacked thymidylate synthase leading to their very narrow antifungal spectrum activity [10].
Echinocandins, including micafungin (12), caspofungin (13), and anidulafungin (14), were comparatively recently discovered as antifungal drugs (Fig. 3) [14]. They were semi-synthetic amphiphilic lipopeptides composed of a cyclic hexapeptide core with different lipid side chains [5, 12, 18]. Echinocandins inhibited the catalytic subunit of 1,3-β-D-glucan synthase (GS), which was responsible for synthesis of 1,3-β-D-glucan, an important component of the fungal cell wall [22]. However, one limitation of the clinically available echinocandins was the need for daily intravenous administration, which was problematic for prolonged therapy [13, 20]. Recently, several new GS inhibitors had been discovered through high-throughput screening of compound libraries from natural products or synthetic molecules, such as pyridazinone (15), enfumafungin (16), and piperazine propanol (17) [16].

Structures of 1,3-β-D-glucan synthase (GS) inhibitors
Several new antifungals are currently in development (Fig. 4). These agents were similar to clinically available antifungal drugs in terms of their mechanisms of action, but were more advantageous than the current drugs, both in terms of overcoming antifungal resistance and avoiding adverse effects [20]. Encouragingly, some of these agents might soon be available for clinical use. These compounds mainly included the following categories: (i) VT-1129 (18), VT-1161 (19), and VT-1598 (21) (fungal-specific inhibitors of Cyp51), which were more specific inhibition of fungal Cyp51 comparing with mammalian CYP450 enzymes [23,24,25]; (ii) AX001 (20, inhibition of fungal glycosylphosphatidylinositol biosynthesis), prevented the maturation of glycosylphosphatidylinositol-anchored proteins by inhibiting inositol acyltransferase [13, 20]; (iii) SCY-078 (22) and CD101 (23) (inhibition of glucan synthase), among, CD101 had a longer half-life ( > 80 h) and SCY-078 allowed for oral administration due to good absorption from the gastrointestinal tract [26,27,28]; (iv) F901318 (24, inhibition of fungal pyrimidine biosynthesis), its activity against A. fumigatus dihydroorotate dehydrogenase was significantly more potent compared with that of the human [29]; (v) T-2307 (25, inducing collapse of fungal mitochondrial membrane potential), it was preferentially taken up by fungal cells compared with mammalian cells [20, 30].

Current antifungals in clinical trials. The chemical structures of VT-1129 (18, fungal-specific inhibitors of Cyp51), VT-1161 (19, fungal-specific inhibitors of Cyp51), AX001 (20, inhibition of fungal glycosylphosphatidylinositol biosynthesis), VT-1598 (21, fungal-specific inhibitors of Cyp51), SCY-078 (22, inhibition of glucan synthase), CD101 (23, inhibition of glucan synthase), F901318 (24, inhibition of fungal pyrimidine biosynthesis), and T-2307 (25, inducing collapse of fungal mitochondrial membrane potential) are shown
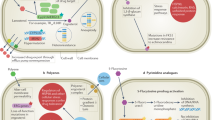
Metabolic targets in the fungal aspartate pathway
The fungal aspartate pathway is required for the biosynthesis of threonine, isoleucine, and methionine (Fig. 5). Pathway is a good target for novel antifungal agents, since elements of this pathway are essential for fungal viability and is not found in mammals [31,32,33,34]. Among them, homoserine transacetylase (MET2), ATP sulfurylase (MET3), transcription factor protein (MET4), methionine synthase (MET6), homocysteine synthase (MET15), aspartate kinase (HOM3), homoserine dehydrogenase (HOM6), homoserine kinase (THR1), threonine synthase (THR4), and acetolactate synthase (ILV2) showed great promise as antifungal targets. Several inhibitors of MET2, MET4, MET15, HOM3, HOM6, THR4, and ILV2 had been discovered till now (Fig. 6).

Ergosterol, methionine, threonine, isoleucine, and valine biosynthetic pathways. a Ergosterol biosynthetic pathway [3]. b Methionine and threonine biosynthetic pathways [31, 35, 38]. c Isoleucine and valine biosynthetic pathways [56]. Only selected substrates or products are shown. Genes that encode enzymes in the pathway are expressed in italics. CoA coenzyme A, MET3 ATP sulfurylase, MET14 adenylyl sulfate kinase, MET16 phosphoadenylyl sulfate reductase, MET5 sulfite reductase-β subunit, MET10 sulfite reductase-α subunit, MET2 homoserine transacetylase, MET15 ( = MET17; MET25), homocysteine synthase (O-acetylhomoserine sulfhydrylase); MET6 methionine synthase, HOM3 aspartate kinase, HOM2 aspartic semi-aldehyde dehydrogenase, HOM6 homoserine dehydrogenase, APS adenylyl sulfate, PAPS phosphoadenylyl sulfate, STR2 cystathionine-γ-synthase, STR3 cystathionine-β-lyase, STR4 cystathionine-β-synthase, STR1 cystathionine-γ-lyase, SAM1/SAM2 S-adenosylmethionine synthase, SUL1/SUL2 sulfate transporter, SAH1 S-adenosylhomocysteine hydrolase, THR1 homoserine kinase, THR4 threonine synthase, ILV1 threonine deaminase, ILV2/ILV6 acetolactate synthase, ILV5 acetohydroxy acid reductoisomerase, ILV3 dihydroxy acid dehydratase, BAT1/BAT2 branched-chain amino acid aminotransferase

Structures of homoserine transacetylase (26), transcription factor protein (27), homocysteine synthase (27), homoserine dehydrogenase (28), aspartate kinase (29, 30, 31, and 32), threonine synthase (33), and acetolactate synthase (34, 35, 36, 37, 38, 39, 40, and 41) inhibitors
Homoserine transacetylase (HTA), encoded by the MET2 gene, catalyzed the transfer of an acetyl group from acetyl-CoA to the hydroxyl group of homoserine. This was a committed step in the biosynthesis of methionine from aspartic acid in many fungi [35,36,37]. Genetic studies in yeast had shown that deletion of the gene that encodes for HTA was lethal in minimal medium [37]. Recent studies showed that met2 mutant was also found to be a methionine auxotroph in Candida guilliermondii and C. neoformans [36, 38]. Moreover, met2 mutant of C. neoformans had been found to be avirulent in the mice model. Fortunately, the first HTA inhibitor CTCQC (6-carbamoyl-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-4-carboxylic acid, 26) had been identified [36]. These studies suggested that HTA was a viable drug target in antifungal research.
MET3 encoded ATP sulfurylase, which converted sulfate to adenylyl sulfate in the inorganic sulfur assimilation [32, 35]. Data in a recent study showed that ATP sulfurylase was essential for the utilization of sulfamate as a sulfur source in the yeast Komagataella pastoris [39]. In C. neoformans, the sulfate-assimilation arm of the methionine biosynthetic pathway was critical for virulence and survival in vivo, and also played an important role in vitro even in the presence of abundant exogenous methionine [32, 33]. Melanin could protect the pathogen against the harsh host environment [31]. Melanin formation was considered as an important virulence factor in C. neoformans [33]. In vitro, the C. neoformans met3 mutant had a substantial defect in melanin formation, and significantly reduced growth rate. In addition, the met3 mutant showed avirulent and weak survivability in the murine inhalation infection model [31, 32]. These studies suggested that MET3 was a promising target for antifungal agents.
Yeast sulfur metabolism was transcriptionally regulated by the activator MET4 [40, 41]. The transcriptional activation of MET2, MET3, MET5, MET10, MET14, MET25 (alternate names: MET15, MET17), and MET16 genes did not occur in met4 mutants [35]. Azoxybacilin (27), produced by Bacillus cereus, had a broad spectrum of antifungal activity in methionine-free medium. These studies showed that azoxybacilin inhibited the transcription regulation of the MET4 gene, which in turn repressed the mRNA syntheses of those genes involved in sulfate assimilation [42]. Azoxybacilin as a gene regulation inhibitor provided a new way of inhibiting fungal cells. Glutathione (GSH) was an important molecule in the protection of yeast cells against damage induced by oxidative stress [43]. It was recently reported that MET4 was also involved in regulation of GSH biosynthesis in the methylotrophic yeast Ogataea (Hansenula) polymorpha [44]. Thus, MET4 was integral to the maintenance of cellular GSH concentrations [43]. As the inorganic sulfur assimilatory pathway was absent in humans, it could be an attractive target for drug designing [31].
MET6 encoded methionine synthase which converted homocysteine to methionine in fungi. The MET6 encoded protein of fungi was a cobalamine (vitamin B12)-independent protein, differing from the human methionine synthase, which was cobalamin-dependent [31, 33]. In C. neoformans, the met6 mutant was a methionine auxotroph, and had been found to be avirulent in the mice inhalation infection model. Moreover, relative to wild-type strains, the met6 mutant grew very slowly and lost viability upon methionine starvation. Capsule production was an important virulence factor, and prevented phagocytosis of C. neoformans by macrophages and neutrophils. However, the met6 mutation resulted in substantially reducing capsule formation. In addition, the deletion of MET6 gene led to toxic accumulation of homocysteine, which consequently resulted in the inhibition of ergosterol biosynthesis [33]. These results suggested that MET6 was an attractive target for antifungal agents.
Homoserine dehydrogenase, encoded by HOM6 gene in Saccharomyces cerevisiae, catalyzed the conversion of aspartate β-semialdehyde to homoserine. It was required for the biosynthesis of threonine, isoleucine, and methionine [45]. The deletion of HOM6 led to toxic accumulation of aspartate β-semialdehyde, which consequently resulted in the inhibition of fungal cell growth [45, 46]. Recent studies had also reported that the deletion of C. albicans HOM6 caused translational arrest in cells grown under amino acid starvation conditions and also led to decline of adhesive capacity of C. albicans [47]. The targeting homoserine dehydrogenase antifungal compound, (s)−2-amino-5-hydroxy-4-oxopentanoic acid (HON; RI-331, 28), which was isolated from Streptomyces species, had been shown to be effective against C. albicans, C. neoformans, and Cladosporium fulvus [48]. Given that there was no HOM6 homolog in mammalian cells, HOM6 could be an excellent target for novel antifungal agents.
The last committed step, the incorporation of sulfide into a carbon chain, was catalyzed by homocysteine synthase (O-acetylhomoserine sulfhydrylase, encoded by MET15) in the inorganic sulfur assimilatory pathway [35]. Met15-deficiency in S. cerevisiae led to a nutritional requirement for methionine, cysteine, or homocysteine [49]. In C. albicans, the deletion of MET15 led to a severe defect of growth on sulfate [31]. The natural product azoxybacilin (27) had been shown to target homocysteine synthase and inhibited the growth of fungi [34]. These studies suggested that MET15 was a potential target for antifungal agents.
Yeast synthesizes threonine from aspartate via five enzymatic reactions steps (Fig. 5) [50]. The first step in this process was activation of aspartate by phosphorylation catalyzed by aspartate kinase (encoded by HOM3) [34]. Previous studies demonstrated that S. cerevisiae hom3 mutant was unable to survive in vivo and C. neoformans hom3 mutant was lethal [51]. From screening a 1000-compounds diversity library, Bareich et al. identified a novel class of 7-chloro-4([1,3,4]thiadiazol−2-ylsulfanyl)-quinoline HOM3 inhibitor (compounds 29, 30, 31, and 32), which had the potential to act as leads in the development of new antifungal agents [34].
Homoserine was converted to threonine by the sequential actions of homoserine kinase (encoded by THR1) and threonine synthase (encoded by THR4) [51]. THR1 and THR4 had been verified to be essential for growth in C. neoformans [52]. In S. cerevisiae, thr1 and thr4 mutants were severely depleted after 4 h in vivo. Similarly, C. albicans thr1 mutants were significantly attenuated in virulence. In addition, S. cerevisiae thr1 and thr4 mutants as well as C. albicans thr1 mutants were extremely serum sensitive. Further studies had shown that low serum threonine concentrations and the accumulation of the biosynthetic intermediate homoserine were key to the rapid death of thr1 and thr4 mutants [51]. Since homoserine kinase and threonine synthase were absent in mammals, they are potential targets for the development of novel antifungal agents. Until now, several threonine synthase inhibitors (rhizocticins, 33) had been discovered [53, 54].
Isoleucine and valine biosynthesis were parallel pathways catalyzed by the same enzymes (Fig. 5) [55]. The isoleucine and valine biosynthetic enzyme acetolactate synthase (encoded by ILV2) catalyzed the conversion of α-ketobutyrate/pyruvate to α-aceto-α-hydroxybutyrate/α-acetolactate. Some studies suggested that S. cerevisiae and C. neoformans ilv2 mutants rapidly lost viability during isoleucine and valine starvation, and did not survive in vivo and/or was avirulent. In addition, the C. albicans ilv2 mutant was significantly attenuated in virulence, and underwent a dramatic decline in viability upon isoleucine and/or valine starvation [56,57,58]. Remarkably, several acetolactate synthase inhibitors (compounds 34, 35, 36, 37, 38, 39, 40, and 41) had been identified. In vitro studies had demonstrated that these compounds possessed good antifungal activity against S. cerevisiae, C. albicans, A. fumigatus, C. neoformans, and Rhizopus oryzae [59].
ROS production
Previous studies demonstrated that oxidative damage induced by endogenous reactive oxygen species (ROS) involved in the antifungal activity of AMB and FLC [60,61,62,63]. ROS were the byproducts of cellular metabolism and primarily generated in the mitochondria [64]. Overproduction of ROS caused serious oxidative stress in the cell and resulted in damage of nucleic acids, lipids, and proteins [65, 66]. Recently, more and more antifungal natural products were discovered with enhancing ROS levels in the cells (Fig. 7). Citronellal (42) was a monoterpenoid isolated from the Cymbopogon plants, and it could increase ROS levels to elicit cell necrosis, mitochondrial dysfunction, and DNA damage in C. albicans [67]. The antifungal natural monoterpenoid perillaldehyde (43), derived from Perilla frutescens, was found to cause accumulation of ROS in C. albicans [68]. HSAF (heat-stable antifungal factor, 44), a polycyclic tetramate macrolactam from Lyaobacter enzymogenes C3, induced the apoptosis of C. albicans through inducing the production of ROS [69]. Retigeric acid B (RAB, 45) from lichen inhibited the growth of C. albicans by stimulating ROS production [64]. In addition, allicin (46)-mediated oxidative damage contributed to the synergistic interaction of allicin and AMB [70]. Thus, oxidative stress responses was an important new targets for antifungal agents discovery.

The inhibitors of potential antifungal targets
Anti-biofilm strategies
In natural environments, most of fungi can from a planktonic to a sessile state forming the so-called biofilms [66]. Fungal biofilms were comprised of adherent cells covered by an extracellular polymeric matrix. More recently, the majority of clinically encountered fungi had been shown to produce biofilms, including Aspergillus, Saccharomyces, Cryptococcus, Candida spp. Among them, the well-studied was C. albicans [71]. Most manifestations of candidiasis were in fact associated with the formation of Candida biofilms [5]. Thus, biofilm formation was considered as an important virulence factor. The proclivity of C. albicans to form biofilms had caused a range of superficial mucosal infections and severe disseminated candidiasis. C. albicans biofilm was intrinsically resistant to the host immune system and conventional antifungal drugs [72]. The resistance of C. albicans biofilm cells to antifungal drugs was higher than that of planktonic cells, and the corresponding MICs were 30–2000 times higher. Therefore, inhibiting biofilm formation was an important for fungal resistance reversing [73].
Previous study showed that fungal prostaglandin (PG) could act as a regulator for biofilm development in C. albicans and that it was also a significant virulence factor in biofilm-associated infections of C. albicans [73, 74]. Prostaglandin E2 (PGE2) belongs to the most abundant PG [75]. C. albicans was known to produce PGE2 from arachidonic acid [76]. Several studies certified that PGE2 could promote fungal cell adhesion, germ tube formation, and biofilm development in C. albicans [76, 77]. Evidence revealed that candidiasis was associated with high levels of PGE2, and decreasing prostaglandin production during C. albican infections was an important factor in relieving chronic infections [73]. Furthermore, PGE2 production was also a significant virulence factor in biofilm-associated infections of non-albicans species [73, 75].
The biosynthesis of PGE2 was catalyzed by several enzymes (Fig. 8). First, arachidonic acid was mediated by cyclooxygenase (COX-1 or COX-2) to form prostaglandin G2 (PGG2), and then, the same enzyme catalyzed the formation of prostaglandin H2 (PGH2). Finally, PGE2 was enzymatically produced from PGH2 via PGE2 synthase [73, 78, 79]. Since PGE2 played an important role in fungal cell adhesion, biofilm development, and germ tube formation, the key enzymes involved in the PGE2 biosynthetic pathway had been attractive targets for antifungal agents. In fact, several studies had suggested that COX inhibitors (e.g., ibuprofen, 47; aspirin, 48; and indomethacin, 49) had strong antifungal activity against C. albicans. These compounds inhibited the biosynthesis of PGE2 by targeting cyclooxygenase, and therefore reduced biofilm development in C. albicans [74, 80, 81]. As the terminal enzyme down-stream of COX-2, microsomal prostaglandin E synthase-1 (mPGES-1) could catalyze the biosynthesis of PGE2 with fewer side effects and, ideally, could not affect the formation of other housekeeping PGs [73]. Thus, mPGES-1 was considered as an attractive antifungal drug target. Fortunately, several compounds were considered to be mPGES-1 inhibitors, such as MF63 (50) and licofelone (51). These compounds could efficiently suppress mPGES-1 activity and reduce PGE2 levels [73, 82].

Prostaglandin E2 (PGE2) biosynthesis pathway based on the arachidonic acid [73]. Only selected substrates or products are shown. Metabolites (green); enzymes (red) that catalyze these reactions in the pathway are shown to the right. PGG2, prostaglandin G2; PGH2, prostaglandin H2; PGE2, prostaglandin E2; COX, cyclooxygenase; mPGES, microsomal prostaglandin E2 synthase
Quorum sensing events were those in which microbial actions and response were correlated to cell density [66]. It has been demonstrated that quorum-sensing molecules were essential for biofilm formation and whose threshold concentration triggered biofilm formation [83]. Farnesol was an extracellular quorum-sensing molecule [84]. When farnesol accumulated above a threshold level, it could prevent C. albicans from converting from the yeast form to the mycelium form to inhibiting biofilms formation. In addition, extra exogenous farnesol also inhibited the translation to limit growth and filamentation in C. albicans and S. cerevisiae [85]. Kovács et al. had shown that farnesol could potentiate the activity of echinocandins (caspofungin, micafungin) against Candida parapsilosis biofilms [84]. Besides the aforementioned effect on morphological transformation, farnesol also affected other biochemical pathways of yeasts, for example, those ones for sterol biosynthesis or triggering of apoptosis via accumulation of ROS to damage essential cellular compartments [86]. More recently, bafilomycin C1 (Baf C1, 52) was isolated from a fermentation broth of Streptomyces albolongus by our research group and it showed strong antifungal activity against C. albicans [87]. Baf C1 also could increase the content of farnesol in C. albicans environment [3]. However, it should be noted that the Baf C1 exhibited significant cytotoxicity. Another study had shown that RAB could induce farnesol production through stimulating the expression of Dpp3p, a protein required for farnesol biosynthesis. The enhanced farnesol induced by RAB probably contributed to the dysfunction of mitochondria and apoptosis in C. albicans [64]. These results suggested that farnesol was a potential target for antifungal agents.
Sulfite transporter as a potential antifungal target
Most superficial fungal infections were caused by dermatophytes, a specialized group of filamentous fungi which exclusively infected keratinized host structures such as hair, skin, or nails, utilizing them as a sole nitrogen and carbon source [88, 89]. Dermatophytes and other filamentous fungi excreted sulfite as a reducing agent during keratin degradation. In the presence of sulfite, cystine in keratin was directly cleaved to cysteine and S-sulphocysteine. Thereby, the reduced proteins became accessible to hydrolysis by various endoproteases and exoproteases secreted by the fungi [90]. Sulfite was produced from cysteine metabolism, and was secreted by dermatophytes and filamentous fungi using a sulfite efflux pump encoded by the gene SSU1. The high expression of SSU1 was characteristic of dermatophytes, which promoted these fungi to efficiently degrade the stratum corneum, hair, and nails [91]. Thus, sulfite transporter SSU1 was considered as one of the most important dermatophyte virulence factors [92]. Several sulfite efflux pumps had been identified in A. fumigatus, Trichophyton rubrum, Arthroderma benhamiae, S. cerevisiae, and C. albicans [90, 93, 94]. It had been proved that A. benhamiae ssu1 mutants were unable to grow specifically on hair and nails [95]. In addition, the deletion of C. albicans SSU1 resulted in enhanced sensitivity of the fungal cells to both cysteine and sulfite. Notably, C. albicans ssu1 mutant displayed reduced hypha formation in the presence of cysteine [94]. Sulfite transporters would be a new target for antifungal drugs in dermatology, since inhibition of these transporters could prevent dermatophytes hydrolyzing keratin. Moreover, these sulfite transporters were absent in humans [31, 90].
Phosphopantetheinyl transferase as a potential antifungal target
The phosphopantetheine (PPT) portion of coenzyme A was an essential group for many carrier proteins and enzymes. The PPT moiety was transferred to a conserved serine residue within the carrier proteins in process of a magnesium ion-dependent reaction catalyzed by phosphopantetheinyl transferases (PPTases) [96, 97]. The majority of filamentous fungi had distinct, functionally diverse, and nonredundant PPTases that could be categorized into three types [98]. The first was integrated within the cytoplasmic fatty acid synthase and transfered the PPT to an acyl carrier protein (ACP) domain within the same protein [97, 99]. The second (Lys5 in S. cerevisiae and C. albicans; PptA in A. fumigatus) PPTase activated α-aminoadipate reductase and was therefore required for lysine biosynthesis [97, 100]. The third PPTase (Ppt2 in S. cerevisiae; PptB in A. fumigatus) was involved in mitochondrial fatty acid synthesis, and was required to transfer the PPT from CoA to the mitochondrial acyl carrier protein Acp1 [99]. In contrast, only a single PPTase was found in humans. Moreover, it was of a different structural class from those in fungi, thereby improving the chance of finding fungal-specific inhibitors [99, 101]. Previous work had shown that phosphopantetheinyl transferase PptB was essential for viability in A. fumigatus [97]. In addition, a recent study also showed that phosphopantetheinyl transferase Ppt2 was essential for growth in C. albicans [99]. In vitro studies have demonstrated that A. fumigatus PptA mutant required supplementation with both lysine and iron to sustain growth and form nonpigmented conidia [97, 98]. In a very recent study, Johns et al. [98] reported PptA was vital for virulence of A. fumigatus. These results suggested that the PptA plays a major role in the production of secondary metabolites, growth under iron-limiting conditions, and virulence of A. fumigatus. Moreover, the PptA inhibitors 6-nitroso-1,2-benzopyrone (53), PD 404,182 (54), and calmidazolium chloride (55) were found to be highly effective against A. fumigatus [98]. These discovery validated phosphopantetheinyl transferase as a potential target of antifungal agents.
Mitochondrial phosphate carrier as a potential antifungal target
Mitochondria had been identified as key players in the adaptive abilities of pathogenic fungi, enabling virulence, morphogenesis, and drug resistance [102,103,104]. Under normal conditions, most of the energy required by cells was supplied by the generation of ATP in their mitochondria. Mitochondrial phosphate carrier proteins performed a critical function in eukaryotes [105]. In S. cerevisiae, the deletion of mitochondrial phosphate carrier Mir1 resulted in delaying growth or failing to grow on non-fermentable substrate [106]. Mir1 had been identified as a structural component of the fungal mitochondrial unselective channel (MUC) [107]. Mir1 manages the transport of inorganic phosphate (Pi) from the cytosol into the inner matrix of fungal mitochondria, where it was used by ATP synthase to generate ATP [105]. In addition, research suggested that Pi itself played an unique role in the mitochondria of fungi. In fungi, the presence of phosphate in the inner mitochondrial matrix was essential for preventing uncoupling of electron transport and loss of membrane potential. This did not appear to be case for mammalian mitochondria [105]. Recently, McLellan et al. [105] had been identified the compound ML316 (56) as an inhibitor of the mitochondrial phosphate carrier Mir1 by using genetic, biochemical, and metabolomic approaches. This compound, distinguished from all previously characterized mitochondrial poisons, selectively inhibited Mir1 resulting in an unusual metabolic catastrophe marked by citrate accumulation. Moreover, ML316 reduced fungal burden and enhanced azole activity in a mouse model of oropharyngeal candidiasis. Thus, targeting Mir1 could provide a new therapeutic strategy [105].
Bromodomain (BD) as a potential antifungal target
The bromodomains and extra-terminal domain (BET) family proteins were chromatin-associated factors that regulated transcription and chromatin remodeling [108, 109]. BET proteins bound chromatin through their two bromodomains (BDs: BD1 and BD2), which recognized specific acetylated histones [110]. In recent years, researchers had shown great interest in the BET family protein targets to discover and develop inhibitors to regulate gene expression by inhibiting the acetylated chromatin interaction [110, 111]. Some studies had demonstrated that small-molecule inhibitors, such as JQ1, I-BET, and I-BET762, which not only had high affinity but also high specificity to BET BDs [110, 112]. These developments made BET family proteins an important therapeutic targets for major diseases such as cancer, neurological disorders, obesity, and inflammation [110].
BET protein BDF1 of S. cerevisiae had been characterized as a global transcriptional regulator, where it regulated over 500 genes. In addition, it had another BET gene BDF2, which was partly functionally redundant with BDF1. Disruption of BDF1 caused severe morphological changes and growth defects, while deletion of both BDF1 and BDF2 was lethal. However, many pathogenic fungal species (including C. albicans, A. fumigatus, and C. neoformans) lacked BDF2, suggesting that inhibition of the sole BET family protein BDF1 might significantly reduce viability and virulence [108]. More recently, Mietton et al. reported that the BDF1 was essential in C. albicans and its mutant presented a loss of viability in vitro and decreasing virulence in vivo. The study also showed that bromodomains BD1 and BD2 in BDF1 of C. albicans could be selectively targeted by small-molecule inhibitors (e.g., dibenzothiazepinone, 57; imidazopyridine, 58) without compromising human BET BD function [108]. These findings laid the foundation for the development of BDF1 BDs inhibitors as a novel class of antifungal drugs.
Conclusion
In the post genomic era, there were plenty of genetic information carried on DNA to use for screening potential drug targets in pathogenic organisms [113]. This will help to find and develop antifungal agents with novel modes of action. Although it was predictable that resistance would eventually develop to antibiotics, it was found that multi-targeting was key for a lower rate of resistance [114]. This could be the developmental direction of antifungal drugs in the future.
Although much progress has been accomplished towards the identification of putative targets that could lead to the development of new antifungal agents, the feasibility and practicality of these new targets remains to be explored.
References
Ikeh M, Ahmed Y, Quinn J. Phosphate acquisition and virulence in human fungal pathogens. Microorganisms. 2017;5:E48 https://doi.org/10.3390/microorganisms5030048.
Gonzalez JM, Rodriguez CA, Agudelo M, Zuluaga AF, Vesga O. Antifungal pharmacodynamics: Latin America’s perspective. Braz J Infect Dis. 2017;21:79–87.
Su H, et al. Bafilomycin C1 exert antifungal effect through disturbing sterol biosynthesis in Candida albicans. J Antibiot (Tokyo). 2018;71:467–476.
Sawant B, Khan T. Recent advances in delivery of antifungal agents for therapeutic management of candidiasis. Biomed Pharmacother. 2017;96:1478–1490.
Rodrigues ME, Silva S, Azeredo J, Henriques M. Novel strategies to fight Candida species infection. Crit Rev Microbiol. 2016;42:594–606.
Pfaller MA, Diekema DJ. Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev. 2007;20:133–63.
Rocha GR, Florez Salamanca EJ, de Barros AL, Lobo CIV, Klein MI. Effect of tt-farnesol and myricetin on in vitro biofilm formed by Streptococcus mutans and Candida albicans. BMC Complement Altern Med. 2018;18:61 https://doi.org/10.1186/s12906-018-2132-x.
Sanglard D, Coste A, Ferrari S. Antifungal drug resistance mechanisms in fungal pathogens from the perspective of transcriptional gene regulation. FEMS Yeast Res. 2009;9:1029–50.
Teodoro GR, Ellepola K, Seneviratne CJ, Koga-Ito CY. Potential use of phenolic acids as anti-Candida agents: a review. Front Microbiol. 2015;6:1420 https://doi.org/10.3389/fmicb.2015.01420.
Campoy S, Adrio JL. Antifungals. Biochem Pharmacol. 2017;133:86–96.
Gavarkar PS, Adnaik RS, Mohite SK. An overview of azole antifungals. Int J Pharm Sci Res. 2013;4:4083–9.
Roemer T, Krysan DJ. Antifungal drug development: challenges, unmet clinical needs, and new approaches. Cold Spring Harb Perspect Med. 2014;4:a019703 https://doi.org/10.1101/cshperspect.a019703.
Wiederhold NP. The antifungal arsenal: alternative drugs and future targets. Int J Antimicrob Agents. 2018;51:333–9.
Prasad R, Shah AH, Rawal MK. Antifungals: mechanism of action and drug resistance. In: Ramos, J, Sychrová, H, Kschischo, M, editors. Yeast membrane transport. Springer: New York, 2016. p. 327–349.
Del Poeta M. Special issue: novel antifungal drug discovery. J Fungi (Basel). 2016;2:33 https://doi.org/10.3390/jof2040033.
Liu N, Tu J, Dong G, Wang Y, Sheng C. Emerging new targets for the treatment of resistant fungal infections. J. Med. Chem. E-pub ahead of print 16 2018; https://doi.org/10.1021/acs.jmedchem.7b01413).
Flick AC, et al. Synthetic approaches to the new drugs approved during 2015. J Med Chem. 2017;60:6480–515.
Odds FC, Brown AJP, Gow NAR. Antifungal agents: mechanisms of action. Trends Microbiol. 2003;11:272–9.
Kathiravan MK, et al. The biology and chemistry of antifungal agents: a review. Bioorg Med Chem. 2012;20:5678–98.
Wiederhold NP. Antifungal resistance: current trends and future strategies to combat. Infect Drug Resist. 2017;10:249–59.
Espinel-Ingroff A. Novel antifungal agents, targets or therapeutic strategies for the treatment of invasive fungal diseases: a review of the literature (2005-9). Rev Iberoam Micol. 2009;26:15–22.
McCarthy MW, Kontoyiannis DP, Cornely OA, Perfect JR, Walsh TJ. Novel agents and drug targets to meet the challenges of resistant fungi. J Infect Dis. 2017;216:S474–S483.
Warrilow AG, et al. The investigational drug VT-1129 is a highly potent inhibitor of Cryptococcus species CYP51 but only weakly inhibits the human enzyme. Antimicrob Agents Chemother. 2016;60:4530–8.
Warrilow AG, et al. The clinical candidate VT-1161 is a highly potent inhibitor of Candida albicans CYP51 but fails to bind the human enzyme. Antimicrob Agents Chemother. 2014;58:7121–7.
Wiederhold NP, et al. Fungal-specific Cyp51 inhibitor VT-1598 demonstrates in vitro activity against Candida and Cryptococcus species, endemic fungi, including Coccidioides species, Aspergillus species and Rhizopus arrhizus. J Antimicrob Chemother. 2018;73:404–8.
James KD, Laudeman CP, Malkar NB, Krishnan R, Polowy K. Structure-activity relationships of a series of echinocandins and the discovery of CD101, a highly stable and soluble echinocandin with distinctive pharmacokinetic properties. Antimicrob Agents Chemother. 2017;61:e01541–16. https://doi.org/10.1128/AAC.01541-16.
Zhao Y, et al. CD101: a novel long-acting echinocandin. Cell Microbiol. 2016;18:1308–16.
Wring SA, et al. Preclinical pharmacokinetics and pharmacodynamic target of SCY-078, a first-in-class orally active antifungal glucan synthesis inhibitor, in murine models of disseminated Candidiasis. Antimicrob Agents Chemother. 2017;61:e02068–16. https://doi.org/10.1128/AAC.02068-16.
Oliver JD, et al. F901318 represents a novel class of antifungal drug that inhibits dihydroorotate dehydrogenase. Proc Natl Acad Sci USA. 2016;113:12809–14.
Nishikawa H, et al. T-2307, a novel arylamidine, is transported into Candida albicans by a high-affinity spermine and spermidine carrier regulated by Agp2. J Antimicrob Chemother. 2016;71:1845–55.
Bachhawat AK, Yadav AK. Metabolic pathways as drug targets: targeting the sulphur assimilatory pathways of yeast and fungi for novel drug discovery. In: Ahmad, I., Owais, M., Shahid, M., Aqil, F, editors. Combating fungal infections: problems and remedy. Springer: New York, 2010. p. 327–346.
Yang Z, Pascon RC, Alspaugh A, Cox GM, McCusker JH. Molecular and genetic analysis of the Cryptococcus neoformans MET3 gene and a met3 mutant. Microbiology. 2002;148:2617–25.
Pascon RC, Ganous TM, Kingsbury JM, Cox GM, McCusker JH. Cryptococcus neoformans methionine synthase: expression analysis and requirement for virulence. Microbiology. 2004;150:3013–23.
Bareich DC, Nazi I, Wright GD. Simultaneous in vitro assay of the first four enzymes in the fungal aspartate pathway identifies a new class of aspartate kinase inhibitor. Chem Biol. 2003;10:967–73.
Thomas D, Surdin-Kerjan Y. Metabolism of sulfur amino acids in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1997;61:503–32.
Nazi I, et al. Role of homoserine transacetylase as a new target for antifungal agents. Antimicrob Agents Chemother. 2007;51:1731–6.
De Pascale G, Nazi I, Harrison PH, Wright GD. β-Lactone natural products and derivatives inactivate homoserine transacetylase, a target for antimicrobial agents. J Antibiot (Tokyo). 2011;64:483–7.
Obando Montoya EJ, et al. Disrupting the methionine biosynthetic pathway in Candida guilliermondii: characterization of the MET2 gene as counter-selectable marker. Yeast. 2014;31:243–51.
Linder T. ATP sulfurylase is essential for the utilization of sulfamate as a sulfur source in the yeast Komagataella pastoris (syn. Pichia pastoris). Curr Microbiol. 2017;74:1021–5.
Carrillo E, et al. Characterizing the roles of Met31 and Met32 in coordinating Met4-activated transcription in the absence of Met30. Mol Biol Cell. 2012;23:1928–42.
Thomas D, Jacquemin I, Surdin-Kerjan Y. MET4, a leucine zipper protein, and centromere-binding factor 1 are both required for transcriptional activation of sulfur metabolism in Saccharomyces cerevisiae. Mol Cell Biol. 1992;12:1719–27.
Aoki Y, et al. Antifungal azoxybacilin exhibits activity by inhibiting gene expression of sulfite reductase. Antimicrob Agents Chemother. 1996;40:127–32.
Wheeler GL, Quinn KA, Perrone G, Dawes IW, Grant CM. Glutathione regulates the expression of gamma-glutamylcysteine synthetase via the Met4 transcription factor. Mol Microbiol. 2002;46:545–56.
Yurkiv, M et al. Gene of the transcriptional activator MET4 is involved in regulation of glutathione biosynthesis in the methylotrophic yeast Ogataea (Hansenula) polymorpha. FEMS Yeast Res. 18. https://doi.org/10.1093/femsyr/foy004 (2018).
Tun NM, et al. Effects of metal ions and hydrogen peroxide on the phenotype of yeasthom6Δ mutant. Lett Appl Microbiol. 2015;60:20–26.
Arévalo-Rodríguez M, Pan X, Boeke JD, Heitman J. FKBP12 controls aspartate pathway flux in Saccharomyces cerevisiae to prevent toxic intermediate accumulation. Eukaryot Cell. 2004;3:1287–96.
Tsai PW, et al. Candida albicans Hom6 is a homoserine dehydrogenase involved in protein synthesis and cell adhesion. J Microbiol Immunol Infect. 2017;50:863–71.
Jacques SL, et al. Enzyme-assisted suicide: molecular basis for the antifungal activity of 5-hydroxy-4-oxonorvaline by potent inhibition of homoserine dehydrogenase. Chem Biol. 2003;10:989–95.
Viaene J, et al. MET15 as a visual selection marker for Candida albicans. Yeast. 2000;16:1205–15.
Kingsbury JM, McCusker JH. Homoserine toxicity in Saccharomyces cerevisiae and Candida albicans homoserine kinase (thr1△) mutants. Eukaryot Cell. 2010;9:717–28.
Kingsbury JM, McCusker JH. Fungal homoserine kinase (thr1△) mutants are attenuated in virulence and die rapidly upon threonine starvation and serum incubation. Eukaryot Cell. 2010;9:729–37.
Kingsbury JM, McCusker JH. Threonine biosynthetic genes are essential in Cryptococcus neoformans. Microbiology. 2008;154:2767–75.
Borisova SA, Circello BT, Zhang JK, van der Donk WA, Metcalf WW. Biosynthesis of rhizocticins, antifungal phosphonate oligopeptides produced by Bacillus subtilis ATCC6633. Chem Biol. 2010;17:28–37.
Gahungu M, et al. Synthesis and biological evaluation of potential threonine synthase inhibitors: Rhizocticin A and Plumbemycin A. Bioorg Med Chem. 2013;21:4958–67.
Ida K, Ishii J, Matsuda F, Kondo T, Kondo A. Eliminating the isoleucine biosynthetic pathway to reduce competitive carbon outflow during isobutanol production by Saccharomyces cerevisiae. Microb Cell Fact. 2015;14:62 https://doi.org/10.1186/s12934-015-0240-6.
Kingsbury JM, McCusker JH. Cytocidal amino acid starvation of Saccharomyces cerevisiae and Candida albicans acetolactate synthase (ilv2△) mutants is influenced by the carbon source and rapamycin. Microbiology. 2010;156:929–39.
Kingsbury JM, Yang Z, Ganous TM, Cox GM, McCusker JH. Cryptococcus neoformans Ilv2p confers resistance to sulfometuron methyl and is required for survival at 37 °C and in vivo. Microbiology. 2004;150:1547–58.
Kingsbury JM, Goldstein AL, McCusker JH. Role of nitrogen and carbon transport, regulation, and metabolism genes for Saccharomyces cerevisiae survival in vivo. Eukaryot Cell. 2006;5:816–24.
Richie DL, et al. Identification and evaluation of novel acetolactate synthase inhibitors as antifungal agents. Antimicrob Agents Chemother. 2013;57:2272–80.
Mahl CD, et al. Induction of ROS generation by fluconazole in Candida glabrata: activation of antioxidant enzymes and oxidative DNA damage. Diagn Microbiol Infect Dis. 2015;82:203–8.
Okamoto Y, Aoki S, Mataga I. Enhancement of amphotericin B activity against Candida albicans by superoxide radical. Mycopathologia. 2004;158:9–15.
Xu Y, et al. Proteomic analysis reveals a synergistic mechanism of fluconazole and berberine against fluconazole-resistant Candida albicans: endogenous ROS augmentation. J Proteome Res. 2009;8:5296–304.
Kobayashi D, et al. Endogenous reactive oxygen species is an important mediator of miconazole antifungal effect. Antimicrob Agents Chemother. 2002;46:3113–7.
Chang WQ, et al. Retigeric acid B exerts antifungal effect through enhanced reactive oxygen species and decreased cAMP. Biochim Biophys Acta. 2011;1810:569–76.
Yun DG, Lee DG. Silymarin exerts antifungal effects via membrane-targeted mode of action by increasing permeability and inducing oxidative stress. Biochim Biophys Acta. 2017;1859:467–74.
Nazzaro F, Fratianni F, Coppola R. Feo, V. Essential Oils and Antifungal Activity. Pharm (Basel). 2017;10:E86 https://doi.org/10.3390/ph10040086.
Saibabu V, Singh S, Ansari MA, Fatima Z, Hameed S. Insights into the intracellular mechanisms of citronellal in Candida albicans: implications for reactive oxygen species-mediated necrosis, mitochondrial dysfunction, and DNA damage. Rev Soc Bras Med Trop. 2017;50:524–9.
Tian H, et al. Calcium and oxidative stress mediate perillaldehyde-induced apoptosis in Candida albicans. Appl Microbiol Biotechnol. 2017;101:3335–45.
Ding Y, et al. HSAF-induced antifungal effects in Candida albicans through ROS-mediated apoptosis. RSC Adv. 2016;6:30895–904.
An M, et al. Allicin enhances the oxidative damage effect of amphotericin B against Candida albicans. Int J Antimicrob Agents. 2009;33:258–63.
Nett JE, Andes D. Fungal Biofilms: In vivo models for discovery of anti-biofilm drugs. Microbiol Spectr. 2015;3:E30 https://doi.org/10.1128/microbiolspec.MB-0008-2014.
Shi W, et al. The combination of minocycline and fluconazole causes synergistic growth inhibition against Candida albicans: an in vitro interaction of antifungal and antibacterial agents. FEMS Yeast Res. 2010;10:885–93.
Liu X, et al. Potential antifungal targets against a Candida biofilm based on an enzyme in the arachidonic acid cascade-a review. Front Microbiol. 2016;7:1925 https://doi.org/10.3389/fmicb.2016.01925.
Alem MA, Douglas LJ. Prostaglandin production during growth of Candida albicans biofilms. J Med Microbiol. 2005;54:1001–5.
Mishra NN, Ali S, Shukla PK. Arachidonic acid affects biofilm formation and PGE2 level in Candida albicans and non-albicans species in presence of subinhibitory concentration of fluconazole and terbinafine. Braz J Infect Dis. 2014;18:287–93.
Ells R, Kemp G, Albertyn J, Kock JL, Pohl CH. Phenothiazine is a potent inhibitor of prostaglandin E2 production by Candida albicans biofilms. FEMS Yeast Res. 2013;13:849–55.
de Matos RF, et al. Nimesulide inhibits pathogenic fungi: PGE2-dependent mechanisms. Folia Microbiol (Praha). 2017;62:169–74.
Park JY, Pillinger MH, Abramson SB. Prostaglandin E2 synthesis and secretion: the role of PGE2 synthases. Clin Immunol. 2006;119:229–40.
Iyer JP, Srivastava PK, Dev R, Dastidar SG, Ray A. Prostaglandin E(2) synthase inhibition as a therapeutic target. Expert Opin Ther Targets. 2009;13:849–65.
Bink A, et al. The nonsteroidal antiinflammatory drug diclofenac potentiates the in vivo activity of caspofungin against Candida albicans biofilms. J Infect Dis. 2012;206:1790–7.
Rusu E, Radu-Popescu M, Pelinescu D, Vassu T. Treatment with some anti-inflammatory drugs reduces germ tube formation in Candida albicans strains. Braz J Microbiol. 2015;45:1379–83.
Côté B, et al. Substituted phenanthrene imidazoles as potent, selective, and orally active mPGES-1 inhibitors. Bioorg Med Chem Lett. 2007;17:6816–20.
Cao YY, et al. cDNA microarray analysis of differential gene expression in Candida albicans biofilm exposed to farnesol. Antimicrob Agents Chemother. 2005;49:584–9.
Kovács R, et al. Effect of caspofungin and micafungin in combination with farnesol against Candida parapsilosisbiofilms. Int J Antimicrob Agents. 2016;47:304–10.
Egbe NE, Dornelles TO, Paget CM, Castelli LM, Ashe MP. Farnesol inhibits translation to limit growth and filamentation in C. albicans and S. cerevisiae. Microb Cell. 2017;4:294–304.
Dižová S, Bujdáková H. Properties and role of the quorum sensing molecule farnesol in relation to the yeast Candida albicans. Pharmazie. 2017;72:307–12.
Ding N, et al. Bafilomycins and odoriferous sesquiterpenoids from Streptomyces albolongus isolated from Elephas maximus feces. J Nat Prod. 2016;79:799–805.
Kröber A, et al. The transcriptional regulators SteA and StuA contribute to keratin degradation and sexual reproduction of the dermatophyte Arthroderma benhamiae. Curr Genet. 2017;63:103–16.
Zaugg C, et al. Gene expression profiling in the human pathogenic dermatophyte Trichophyton rubrum during growth on proteins. Eukaryot Cell. 2009;8:241–50.
Léchenne B, et al. Sulphite efflux pumps in Aspergillus fumigatus and dermatophytes. Microbiology. 2007;153:905–13.
Baldo A, et al. Mechanisms of skin adherence and invasion by dermatophytes. Mycoses. 2012;55:218–23.
Kasperova A, Kunert J, Raska M. The possible role of dermatophyte cysteine dioxygenase in keratin degradation. Med Mycol. 2013;51:449–54.
Avram D, Bakalinsky AT. SSU1 encodes a plasma membrane protein with a central role in a network of proteins conferring sulfite tolerance in Saccharomyces cerevisiae. J Bacteriol. 1997;179:5971–4.
Hennicke F, et al. Factors supporting cysteine tolerance and sulfite production in Candida albicans. Eukaryot Cell. 2013;12:604–13.
Grumbt M, et al. Keratin degradation by dermatophytes relies on cysteine dioxygenase and a sulfite efflux pump. J Invest Dermatol. 2013;133:1550–5.
Lambalot RH, et al. A new enzyme superfamily-the phosphopantetheinyl transferases. Chem Biol. 1996;3:923–36.
Allen G, et al. Functional analysis of a mitochondrial phosphopantetheinyl transferase (PPTase) gene pptB in Aspergillus fumigatus. Fungal Genet Biol. 2011;48:456–64.
Johns A, et al. A nonredundant phosphopantetheinyl transferase, PptA, is a novel antifungal target that directs secondary metabolite, siderophore, and lysine biosynthesis in Aspergillus fumigatus and is critical for pathogenicity. mBio. 2017;8:e01504–16. https://doi.org/10.1128/mBio.01504-16.
Dobb KS, et al. Characterisation of the Candida albicans phosphopantetheinyl transferase Ppt2 as a potential antifungal drug target. PLoS One. 2015;10:e0143770 https://doi.org/10.1371/journal.pone.0143770.
Guo S, Bhattacharjee JK. Molecular characterization of the Candida albicans LYS5 gene and site-directed mutational analysis of the PPTase (Lys5p) domains for lysine biosynthesis. FEMS Microbiol Lett. 2003;224:261–7.
Joshi AK, Zhang L, Rangan VS, Smith S. Cloning, expression, and characterization of a human 4’-phosphopantetheinyl transferase with broad substrate specificity. J Biol Chem. 2003;278:33142–9.
Sun N, et al. Azole susceptibility and transcriptome profiling in Candida albicans mitochondrial electron transport chain complex I mutants. Antimicrob Agents Chemother. 2013;57:532–42.
Shingu-Vazquez M, Traven A. Mitochondria and fungal pathogenesis: drug tolerance, virulence, and potential for antifungal therapy. Eukaryot Cell. 2011;10:1376–83.
Grahl N, et al. Mitochondrial activity and Cyr1 are key regulators of Ras1 activation of C. albicans virulence pathways. PLoS Pathog. 2015;11:e1005133 https://doi.org/10.1371/journal.ppat.1005133.
McLellan CA, et al. Inhibiting mitochondrial phosphate transport as an unexploited antifungal strategy. Nat Chem Biol. 2018;14:135–41.
Seifert EL, Ligeti E, Mayr JA, Sondheimer N, Hajnóczky G. The mitochondrial phosphate carrier: Role in oxidative metabolism, calcium handling and mitochondrial disease. Biochem. Biophys Res Commun. 2015;464:369–75.
Gutiérrez-Aguilar M, Pérez-Martínez X, Chávez E, Uribe-Carvajal S. Saccharomyces cerevisiae, the phosphate carrier is a component of the mitochondrial unselective channel. Arch Biochem Biophys. 2010;494:184–91.
Mietton F, et al. Selective BET bromodomain inhibition as an antifungal therapeutic strategy. Nat Commun. 2017;8:15482 https://doi.org/10.1038/ncomms15482.
Sahni JM, Keri RA. Targeting bromodomain and extraterminal proteins in breast cancer. Pharmacol Res. 2018;129:156–76.
Padmanabhan B, Mathur S, Manjula R, Tripathi S. Bromodomain and extra-terminal (BET) family proteins: New therapeutic targets in major diseases. J Biosci. 2016;41:295–311.
Ramadoss M, Mahadevan V. Targeting the cancer epigenome: synergistic therapy with bromodomain inhibitors. Drug Discov Today. 2018;23:76–89.
Leal AS, et al. Bromodomain inhibitors, JQ1 and I-BET 762, as potential therapies for pancreatic cancer. Cancer Lett. 2017;394:76–87.
Tripathi H, Luqman S, Meena A, Khan F. Genomic identification of potential targets unique to Candida albicans for the discovery of antifungal agents. Curr Drug Targets. 2014;15:136–49.
Singh SB, Young K, Silver LL. What is an “ideal” antibiotic? Discovery challenges and path forward. Biochem Pharmacol. 2017;133:63–73.
Acknowledgements
This work was funded by the Fundamental Research Funds for the Central Universities, China (No. N172004004).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Su, H., Han, L. & Huang, X. Potential targets for the development of new antifungal drugs. J Antibiot 71, 978–991 (2018). https://doi.org/10.1038/s41429-018-0100-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-018-0100-9
This article is cited by
-
Design, Synthesis and Antifungal Activity of Phloroglucinol Derivatives
Pharmaceutical Chemistry Journal (2022)
-
A semisynthetic borrelidin analogue BN-3b exerts potent antifungal activity against Candida albicans through ROS-mediated oxidative damage
Scientific Reports (2020)
-
Anti-fungal properties and mechanisms of melittin
Applied Microbiology and Biotechnology (2020)
