
Abstract
Using genome mining, a new cytotoxic peptide named curacozole was isolated from Streptomyces curacoi. Through ESI-MS and NMR analyses, curacozole was determined to be a macrocyclic peptide containing two isoleucine, two thiazole and three oxazole moieties. Curacozole exhibited potent cytotoxic activity against HCT116 and HOS cancer cells. The proposed biosynthetic gene cluster of curacozole was identified and compared with that of the related compound YM-216391.
Similar content being viewed by others


Introduction
A large collection of bacterial genome sequences have been accumulated in a database, and genome mining has been used to find new secondary metabolites by utilizing the genomic sequence data [1,2,3]. The biosynthetic gene clusters (BGCs) of ribosomally synthesized and post-translationally modified peptides (RiPPs), including lantibiotics, lasso peptides, and thiopeptides, are suitable targets for identification by the genome mining method since the structural prediction of the final products is comparatively easier than that for peptides biosynthesized by nonribosomal peptide synthetases (NRPSs). The prediction software antiSMASH was developed to search for secondary metabolite BGCs in whole genome sequences [4]. An increase in the understanding the biosynthetic systems of natural products has improved the ability to predict the possible biosynthetic gene clusters for secondary metabolites such as polyketides, NRPs, and RiPPs from the genomic data of bacteria and fungi [5,6,7,8]. Recently, a new prediction system for RiPPs named RODEO (Rapid ORF Description and Evaluation Online) was developed by combining the hidden-Markov-model-based analysis, heuristic scoring, and machine learning to identify biosynthetic gene clusters and predict RiPP precursor peptides [9]. This approach enabled the comprehensive mapping of RiPP biosynthetic gene clusters, revealing the existence of more than 1,300 putative compounds. Based on this bioinformatic approach, six new lasso peptides were discovered and characterized, indicating that the RODEO system is efficient [9].
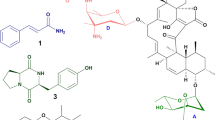
Among RiPPs, telomestatin, a macrocyclic compound containing seven oxazoles and one thiazole, was isolated from Streptomyces anulatus as a potent telomerase inhibitor [10]. Due to its structural characteristics, telomestatin interacts with human telomeric intramolecular G-quadruplexes to form basket-type G-quadruplex (G4) structures from hybrid-type G-quadruplexes [11]. The telomestatin BGC was previously identified by analyzing the genome of Streptomyces anulatus 3533-SV4, which indicated that biosynthesis of telomestatin belonged to the RiPP biosynthetic system [12]. The analogous macrocyclic compound, YM-216391 (2 in Fig. 1), was isolated as a cytotoxic compound from Streptomyces nobilis [13, 14]. The BGC of YM-216391 (2 in Fig. 1) was determined from the genomic sequence data of S. nobilis JCM 4274 [15]. The gene cluster consists of a precursor peptide coding gene and modification genes, which typically compose RiPP BGCs.

Chemical structure of 1 and 2, bold letters around compounds indicate precursor amino acids (FIIGSTCC for 1 and FIVGSSSC for 2)
Recently, the antibacterial peptide curacomycin was isolated from Streptomyces curacoi via genome mining [16]. By further mining the genome of S. curacoi, we found a gene cluster analogous to that of YM-216391 (2 in Fig. 1). As a result of our investigation, a new cytotoxic compound named curacozole (1 in Fig. 1) was isolated from S. curacoi, and its structure was determined by ESI-MS and NMR experiments. Here, we describe the isolation and structure determination of curacozole (1).
Results and discussion
The cytotoxic compound YM-216391 (2 in Fig. 1) was reported to be biosynthesized from the precursor peptide (AFJ68074.1) via several enzymatic modifications [15]. Through modification, the amino acids Ser and Cys in the core peptide sequence FIVGSSSC (bold letters in Fig. 1 and underlined letters in Fig. 2) are converted to oxazoles and thiazoles. By performing a BLASTp search [17] using the amino acid sequence of the precursor peptide of YM-216391 (AFJ68074.1) [15], we found similar precursor peptide coding genes (Fig. 2) in the genomes of S. curacoi DSM 40107T and Streptomyces viridochromogenes Tü57 [3], along with another unique precursor peptide gene in the Streptomyces aurantiacus JA 4570 genome. The new peptide was expected to possess the structure depicted in Fig. 1 as 1, and it may be biosynthesized from the core peptide sequence FIIGSTCC (bold letters in Fig. 1 and underlined letters in Fig. 2). We performed HPLC and ESI-MS investigations on a MeOH extract of S. curacoi NBRC 12761T cells to detect the expected peptide (1) shown in Fig. 1. The expected peptide was detected by the combination of HPLC and ESI-MS (data not shown).

Amino acid sequences of precursor peptide-coding genes for 1 and 2, bold letters indicate conserved amino acids. Underlines indicate core peptide regions. Accession number of each gene is following Streptomyces nobilis: AFJ68074.1, S. curacoi: WP_107116988.1, S. viridochromogenes: WP_003997107.1, S. aurantiacus: WP_016640788.1
To increase the yield of 1 in S. curacoi, rifampicin was used according to the previously reported method [18]. Streptomyces strains with a specific RNA polymerase β gene (rpoB) mutation that confers rifampicin resistance can produce abundant quantities of secondary metabolites [19, 20]. Briefly, the rifampicin-resistant (rifr) S. curacoi mutants were obtained for curacozole production screening. Then, cell extracts of the rifr mutants (30 strains) were analyzed by HPLC to evaluate curacozole production. We found that three rifampicin-resistant mutants harboring a 1298 C > T (Ser433Leu: S433L) mutation in the gene encoding the RNA polymerase β subunit showed a productions of 1 3–5 times higher compared with that of the wild type (data not shown). In this study, we found that the curacozole-overproducing rifr S. curacoi mutants all carried an S433L mutation in the rpoB gene. Previous studies showed that a Streptomyces lividans strain with a single S433L mutation in the rpoB gene overproduced the blue-pigmented antibiotic actinorhodin [21]. These findings indicate that the rpoB S433L mutation effectively improves secondary metabolite production in a variety of streptomycetes. To produce 1, we cultured a rifr mutant designated R25.
To obtain a sufficient quantity of 1 for structure determination, S. curacoi R25 was cultured in 2 L of ISP2 agar media [22]. After 7 days of cultivation, spore and aerial hyphae cells were harvested and extracted with MeOH and then centrifuged to remove any insoluble compounds. The extract was concentrated by rotary evaporation and subjected to a CHP-20P column eluted with 10%, 60%, and 100% MeOH. The 100% MeOH fraction was repeatedly subjected to HPLC purification to afford 1.
The molecular formula of 1 was established to be C36H36N8O6S2 by accurate mass analysis since the ion corresponding to [M + H]+ (the calculated m/z value, 741.2277) was observed at m/z 741.2284. To obtain further information on the chemical structure, NMR experiments including 1H, 13C, DEPT-135, DQF-COSY, HSQC, TOCSY and HMBC analyses of 1 were performed using DMSO-d6 as the solvent (Table 1 and Fig. 3). The 1H NMR spectrum of 1 exhibited 34 proton signals, including three amide protons, one aminomethylene, and two aminomethines. The HSQC experiment direct 1H-13C connections. By analyzing the TOCSY and HMBC spectra, the structure of 1 was established as shown in Fig. 3. Since 1 was expected to be an analog of YM-216391, their NMR spectral data were compared. As shown in Table 1, the chemical shift values of 1 were similar to those of 2. There are three major differences in the NMR spectrum of 1 compared with that of 2. The characteristic chemical shift of position 31 (δH 8.60, δC 120.8) indicated the presence of a thiazole group as opposed to an oxazole group. HMBC correlations from the methyl residue (H28) to C25, C26, C29 and C23 were observed, which indicated that the methyl is attached to a carbon (C25). The H-H spin system constructed by TOCSY and HMBC correlations from H14 to C13 and C11 indicated the presence of an Ile containing C14. The connections among C39, C41, and C48 were not established due to the absence of an HMBC correlation; however, the similarity of the chemical shifts (Table 1) indicated the presence of an oxazole group.

Selected 2D NMR correlations of 1
The absolute stereochemistries of the two Ile groups were analyzed by the modified Marfey’s method [23], and chiral HPLC analysis was performed on the acid hydrolysate of 1. The modified Marfey’s analysis revealed the presence of L-Ile and that the mixture contained D-Ile and D-allo-Ile in a 1:1 molar ratio. The D-Ile and D-allo-Ile moieties could not be distinguished due to their identical HPLC retention times in the modified Marfey’s analysis. To determine the proportions of d-Ile and d-allo-Ile, chiral HPLC analysis was performed on the hydrolysate of 1 according to the previous report [14]. The analysis revealed the presence of d-allo-Ile and d-Ile in a 6:4 molar ratio.
To investigate whether treatment with 1 alters the proliferation of cancer cells, we incubated HCT116 and HOS cells with various concentrations of 1 for 72 h and assessed cell viability by the CellTiter-Glo luminescent cell viability assay. Treatment with 1 resulted in a dose-dependent cytotoxicity in these cell lines and exhibited IC50 values of 8.6 nM and 10.5 nM for HCT116 and HOS cells, respectively (data not shown). These data indicate that 1 is highly toxic towards HCT116 and HOS cancer cells.
In this study, we found a new precursor peptide gene similar to that of YM-216391 in the S. curacoi genome by genome mining (Fig. 2). We successfully isolated the product, named curacozole (1), from the strain. Thus, the cluster containing the precursor peptide gene was considered to be responsible for the synthesis of 1. This gene cluster includes ten genes (Fig. 4) and shares a similar gene organization to that of YM-216391 [15]. Nine of the ten genes, with the exception of AQI70_RS14565, are orthologues of ymI, ymA, ymD, ymE, ymB1, ymC1, ymF, ymBC and ymR1, genes responsible for the biosynthesis of YM-216391 [15]. The translated amino acid sequences of the genes showed 69 to 97% similarity to Ym proteins (Table S1). In light of the structural similarity between 1 and 2 (Fig. 1) and the YM-216391-biosynthetic pathway [15], we propose the biosynthetic pathway of 1 (Fig. 5). A linear precursor peptide of 44 amino acid residues (AQI70_RS38515, Fig. 2) is ribosomally synthesized. A docking protein (AQI70_RS14600) and a cyclodehydratase-oxidoreductase didomain enzyme (AQI70_RS37415) convert G-S-T-C-C into the Gly-oxazole-methyloxazole-thiazole-thiazole moiety. The P-450 (AQI70_RS14595) hydroxylates the Phe residue in the intermediate peptide. The protease (AQI70_RS14580) cleaves the core peptide, which corresponds to 1, at both the N- and C-termini of the full-length precursor, which is followed by N-C terminal cyclization. This cyclic peptide formation may require the assistance of a hypothetical protein (AQI70_RS14605). The cyclodehydratase (AQI70_RS14590) cyclizes thiazole-β-hydroxy-Phe, and then the FAD-binding monooxygenase (AQI70_RS14585) oxidizes the ring to form a thiazole-phenyloxyazole moiety. Although, one of the two Ile residues in 1 may be d-Ile/d-allo-Ile, as this cluster does not include an epimerase homologous to YmG. The epimerase responsible for the conversion of l-Ile into d-Ile/d-allo-Ile still remains to be identified. In cypemycin biosynthesis, CypI is proposed to isomerize L-Ile residues [24]. The S. curacoi genome encodes a homolog of CypI (AQI70_17525) at a locus distant from the cluster, which may catalyze the isomerization.

Biosynthetic gene cluster for curacozole (1). The genes AQI70_RS14605, AQI70_RS38515, AQI70_RS14600, AQI70_RS14595, AQI70_RS14590, AQI70_RS14585, AQI70_RS14580, AQI70_RS37415 and AQI70_RS14560 encode orthologues for ymI, ymA, ymD, ymE, ymB1, ymC1, ymF, ymBC and ymR1, respectively. Accession numbers of the genes are following; AQI70_RS14605:WP_062148887, AQI70_RS38515:WP_107116988, AQI70_RS14600:WP_079051299, AQI70_RS14580:WP_062148872, AQI70_RS37415:WP_062148869, AQI70_RS14565:WP_062148866, AQI70_RS14560:WP_062148863.1

Biosynthetic pathway of curacozole (1)
Recently, genome mining has often been employed to search for RiPP compounds such as lantibiotics, lasso peptides and thiopeptides [25, 26]. Nevertheless, to the best of our knowledge, this is the first report on the genome mining-based discovery of a new sequential oxazole/methyloxazole/thiazole ring-containing macrocyclic peptide from actinomycetes. This may be due to the fact that few compounds of this class of peptides have been reported to date [12, 15]. Investigation of these compounds and their BGCs will contribute to the acceleration of secondary metabolite discovery in the post-genomic era.
Materials and methods
Microbial strains
The bacterial strain Streptomyces curacoi NBRC 12761T was obtained from the NBRC culture collection (NITE Biological Resource Center, Japan). Spontaneous rifampicin-resistant (rifr) mutants of S. curacoi NBRC 12761 were obtained from colonies grown within 10 days after the spores or hyphal fragments (approximately 2 × 109 colony forming units) were spread on GYM agar medium [27] containing 10 µg/mL of rifampicin, which corresponds to an amount that is 2-fold greater than the minimum inhibitory concentration.
Mutation analysis of the rpoB gene
The partial rpoB gene fragment in rifr mutants was obtained using the primers 5′-GGCGCTCGGCTGGACGACCG-3′ (forward) and 5′-CGATCAGACCGATGTTCGGG-3′ (reverse), which were designed based on the sequence in S. curacoi NBRC 12761T. PCR amplification was carried out with Tks Gflex DNA polymerase (TaKaRa Bio, Inc., Shiga, Japan). The purified PCR product was subjected to DNA sequence analysis, which was performed by Eurofins Genomics K. K. (Tokyo, Japan). The sequence data were aligned using the ClustalW program at DDBJ (http://clustalw.ddbj.nig.ac.jp/).
Isolation of curacozole
The Streptomyces curacoi mutant strain R25 was cultured in 2 L of ISP2 agar medium [22] at 30 °C for 7 days. The aerial hyphae and spore cells were harvested and then extracted with MeOH. Approximately 200 ml of MeOH was added to the harvested cells, and the extract was filtered through filter paper (Whatman No.1, GE Healthcare Life Sciences, Little Chalfont, UK). The extract was concentrated to an aqueous residue by a rotary evaporator. The concentrated extract was subjected to open column chromatography (styrene-divinylbenzene resin, CHP-20P, Mitsubishi Chemical Corp., Tokyo, Japan) eluted with 10% MeOH, 60% MeOH and 100% MeOH. The 100% MeOH fraction was concentrated on a rotary evaporator and then separated via HPLC using an ODS column (4.6 × 250 mm, Wakopak Handy ODS, WAKO). The UV detector of HPLC was set at a wavelength of 220 nm. 100% MeOH fraction was subjected to HPLC with elution of 73% MeCN containing 0.05% TFA (trifluoroacetic acid) at flow rate of 1 mL/min to yield 4.0 mg of curacozole (retention time; 12.0 min).
MS experiments
ESI-MS analyses were performed using a JEOL JMS-T100LP mass spectrometer. For accurate MS analysis, reserpine was used as an internal standard.
NMR experiments
An NMR sample was prepared by dissolving 1 in 500 μl of DMSO-d6. All NMR spectra were obtained on Bruker Avance 600 and Avance III HD 800 spectrometers with quadrature detection in the phase-sensitive mode by States-TPPI (time-proportional phase incrementation) and in the echo-antiecho mode. One-dimensional (1D) 1H, 13C, and DEPT-135 spectra were recorded at 25 °C with a 12 ppm window for proton and 239 ppm or 222 ppm windows for carbon. The following 2D 1H-NMR spectra were recorded at 25 °C with 12 ppm or 15 ppm spectral widths in the t1 and t2 dimensions: 2D double-quantum filtered correlated spectroscopy (DQF-COSY), recorded with 512 and 1024 complex points in the t1 and t2 dimensions; 2D homonuclear total correlated spectroscopy (TOCSY) with MLEV-17 mixing sequence, recorded with mixing time of 80 ms, 256 and 1024 complex points in t1 and t2 dimensions; 2D nuclear Overhauser effect spectroscopy (NOESY), recorded with mixing time of 200 and 400 ms, 256 and 1024 complex points in the t1 and t2 dimensions. 2D 1H-13C heteronuclear single quantum correlation (HSQC) and heteronuclear multiple bond connectivity (HMBC) spectra were acquired at 25 °C in the echo-antiecho mode. The 1H-13C HSQC and HMBC spectra were recorded with 1024 × 512 complex points for 12 ppm in the 1H dimension and 160 ppm or 222 ppm in the 13C dimension, respectively, at a natural isotope abundance.
All NMR spectra were processed using TOPSPIN 3.5 (Bruker). Before Fourier transformation, the shifted sinebell window function was applied to the t1 and t2 dimensions. All 1H and 13C dimensions were referenced to DMSO-d6 at 25 °C.
Modified Marfey’s analysis
Compound 1 was subjected to acid hydrolysis with 6N HCl at 110 °C for 16 h. The hydrolysate was concentrated to dryness using a rotary evaporator, and then 200 μL of water was added. To the hydrolysate, 10 μL of a solution of Nα-(5-fluoro-2,4-dinitrophenyl)-l-leucinamide (l-FDLA, Tokyo Chemical Industry Co., LTD, Tokyo, Japan) in acetone (10 μg/μL) and 100 μL of 1 M NaHCO3 solution were added,and the mixture was incubated at 80 °C for 3 min. The reaction mixture was cooled down at room temperature before it was neutralized with 50 μL of 2N HCl and diluted with 1 mL of 50% MeCN. For the standard amino acids, each amino acid was derivatized with l-FDLA and d-FDLA using the same method. Approximately 30 μL of each FDLA derivative was subjected to HPLC analysis on a C18 column (4.6 × 250 mm, Wakopak Handy ODS, Wako). A DAD (MD-2018, JASCO, Tokyo, Japan) was used for the detection of the amino acid derivatives using the absorbance data from 220 nm to 420 nm. The HPLC analysis was performed at a flow rate of 1 mL/min using solvent A (distilled water containing 0.05% TFA) and solvent B (MeCN containing 0.05%TFA) with a linear gradient program from 0 min to 70 min and increasing the percentage of solvent B from 25% to 60%. The retention times (min) of l- and d-FDLA derivatized amino acids in this HPLC condition were the following; l-allo-Ile-l-FDLA (41.20 min), l-Ile-l-FDLA (42.22 min), l-Ile-d-FDLA (58.16 min) and l-allo-Ile-d-FDLA (58.16 min).
Chiral HPLC Analysis
The hydrolysate of the peptide was analyzed by chiral HPLC on a SUMICHIRAL OA5000 (150 × 4.6 mm, Sumika Chemical Analyervice). The HPLC analysis was performed with UV detection at 254 nm using 2 mM CuSO4 containing 5% MeCN as a mobile phase and a flow rate of 1 mL/min. The retention time of the amino acids in this HPLC condition were the following; d-allo-Ile (12.72 min) and d-Ile (15.90 min).
Cytotoxic assay
HCT116 and HOS cells (2.5 × 103) were aliquoted in 96-well plates and treated with 1 (5–100 nM) in D-MEM (HCT116) or E-MEM (HOS) containing FBS (10%). Cell viability was assayed after 72 h by using the CellTiter-Glo luminescent cell viability assay (Promega, Madison, USA) with a JNR Luminescencer (ATTO, Tokyo, Japan) according to the manufacturer’s protocol.
References
Li L, Jiang W, Lu Y. New strategies and approaches for engineering biosynthetic gene clusters of microbial natural products. Biotechnol Adv. 2017;35:936–49.
Velasquez JE, van der Donk WA. Genome mining for ribosomally synthesized natural products. Curr Opin Chem Biol. 2011;15:11–21.
Ju KS, et al. Discovery of phosphonic acid natural products by mining the genomes of 10,000 actinomycetes. Proc Natl Acad Sci USA. 2015;112:12175–80.
Medema MH, et al. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011;39:W339–46.
Blin K, et al. antiSMASH 2.0--a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013;41:W204–12.
Blin K, Kazempour D, Wohlleben W, Weber T. Improved lanthipeptide detection and prediction for antiSMASH. PLoS ONE. 2014;9:e89420.
Weber T, et al. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43:W237–43.
Blin K, Medema MH, Kottmann R, Lee SY, Weber T. The antiSMASH database, a comprehensive database of microbial secondary metabolite biosynthetic gene clusters. Nucleic Acids Res. 2017;45:D555–9.
Tietz JI, et al. A new genome-mining tool redefines the lasso peptide biosynthetic landscape. Nat Chem Biol. 2017;13:470–8.
Shin-ya K, et al. Telomestatin, a novel telomerase inhibitor from Streptomyces anulatus. J Am Chem Soc. 2001;123:1262–3.
Kim MY, Vankayalapati H, Shin-ya K, Wierzba K, Hurley LH. Telomestatin, a potent telomerase inhibitor that interacts quite specifically with the human telomeric intramolecular G-quadruplex. J Am Chem Soc. 2002;124:2098–9.
Amagai K, et al. Identification of a gene cluster for telomestatin biosynthesis and heterologous expression using a specific promoter in a clean host. Sci Rep. 2017;7:3382.
Sohda K, Nagai K, Yamori T, Suzuki K, Tanaka A. YM-216391, a novel cytotoxic cyclic peptide from Streptomyces nobilis. I. fermentation, isolation and biological activities. J Antibiot. 2005;58:27–31.
Sohda K, Hiramoto M, Suzumura K, Takebayashi Y, Suzuki K, Tanaka A. YM-216391, a novel cytotoxic cyclic peptide from Streptomyces nobilis. II. Physico-chemical properties and structure elucidation. J Antibiot. 2005;58:32–6.
Jian XH, et al. Analysis of YM-216391 biosynthetic gene cluster and improvement of the cyclopeptide production in a heterologous host. ACS Chem Biol. 2012;7:646–51.
Kaweewan I, Komaki H, Hemmi H, Kodani S. Isolation and structure determination of new antibacterial peptide curacomycin based on genome mining. Asian J Org Chem. 2017;6:1838–44.
Morgulis A, Coulouris G, Raytselis Y, Madden TL, Agarwala R, Schaffer AA. Database indexing for production MegaBLAST searches. Bioinformatics. 2008;24:1757–64.
Hosaka T, et al. Antibacterial discovery in actinomycetes strains with mutations in RNA polymerase or ribosomal protein S12. Nat Biotechnol. 2009;27:462–4.
Ochi K, Hosaka T. New strategies for drug discovery: activation of silent or weakly expressed microbial gene clusters. Appl Microbiol Biotechnol. 2013;97:87–98.
Thong WL, Shin-ya K, Nishiyama M, Kuzuyama T. Methylbenzene-containing polyketides from a Streptomyces that spontaneously acquired rifampicin resistance: structural elucidation and biosynthesis. J Nat Prod. 2016;79:857–64.
Hu H, Zhang Q, Ochi K. Activation of antibiotic biosynthesis by specified mutations in the rpoB gene (encoding the RNA polymerase beta subunit) of Streptomyces lividans. J Bacteriol. 2002;184:3984–91.
Shirling EB, Gottlieb D. Methods for characterization of Streptomyces species. Int J Syst Bacteriol. 1966;16:313–40.
Harada K, Fujii K, Hayashi K, Suzuki M, Ikai Y, Oka H. Application of D, L-FDLA derivatization to determination of absolute configuration of constituent amino acids in peptide by advanced Marfey’s method. Tetrahedron Lett. 1996;37:3001–4.
Claesen J, Bibb M. Genome mining and genetic analysis of cypemycin biosynthesis reveal an unusual class of posttranslationally modified peptides. Proc Natl Acad Sci USA. 2010;107:16297–302.
Kodani S, Komaki H, Ishimura S, Hemmi H, Ohnishi-Kameyama M. Isolation and structure determination of a new lantibiotic cinnamycin B from Actinomadura atramentaria based on genome mining. J Ind Microbiol Biotechnol. 2016;43:1159–65.
Kaweewan I, Komaki H, Hemmi H, Kodani S. Isolation and structure determination of a new thiopeptide globimycin from Streptomyces globisporus subsp. globisporus based on genome mining. Tetrahedron Lett. 2018;59:409–14.
Shima J, Hesketh A, Okamoto S, Kawamoto S, Ochi K. Induction of actinorhodin production by rpsL (encoding ribosomal protein S12) mutations that confer streptomycin resistance in Streptomyces lividans and Streptomyces coelicolor A3(2). J Bacteriol. 1996;178:7276–84.
Acknowledgements
This study was supported by the Japan Society for the Promotion of Science by Grants-in-aids (grant number 16K01913). The research was partly supported by the Sasakawa Scientific Research Grant from The Japan Science Society (grant number 2018-3001). The NMR spectra were recorded on Bruker Avance 600 and Avance III HD 800 spectrometers at Advanced Analysis Center, NARO.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Kaweewan, I., Komaki, H., Hemmi, H. et al. Isolation and structure determination of a new cytotoxic peptide, curacozole, from Streptomyces curacoi based on genome mining. J Antibiot 72, 1–7 (2019). https://doi.org/10.1038/s41429-018-0105-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-018-0105-4
This article is cited by
-
A putative mechanism underlying secondary metabolite overproduction by Streptomyces strains with a 23S rRNA mutation conferring erythromycin resistance
Applied Microbiology and Biotechnology (2020)
-
Lincomycin-Induced Secondary Metabolism in Streptomyces lividans 66 with a Mutation in the Gene Encoding the RNA Polymerase Beta Subunit
Current Microbiology (2020)
