
Abstract
Zhu–Tokita–Takenouchi–Kim (ZTTK) syndrome, an intellectual disability syndrome first described in 2016, is caused by heterozygous loss-of-function variants in SON. Its encoded protein promotes pre-mRNA splicing of many genes essential for development. Whereas individual phenotypic traits have previously been linked to erroneous splicing of SON target genes, the phenotypic spectrum and the pathogenicity of missense variants have not been further evaluated. We present the phenotypic abnormalities in 52 individuals, including 17 individuals who have not been reported before. In total, loss-of-function variants were detected in 49 individuals (de novo in 47, inheritance unknown in 2), and in 3, a missense variant was observed (2 de novo, 1 inheritance unknown). Phenotypic abnormalities, systematically collected and analyzed in Human Phenotype Ontology, were found in all organ systems. Significant inter-individual phenotypic variability was observed, even in individuals with the same recurrent variant (n = 13). SON haploinsufficiency was previously shown to lead to downregulation of downstream genes, contributing to specific phenotypic features. Similar functional analysis for one missense variant, however, suggests a different mechanism than for heterozygous loss-of-function. Although small in numbers and while pathogenicity of these variants is not certain, these data allow for speculation whether de novo missense variants cause ZTTK syndrome via another mechanism, or a separate overlapping syndrome. In conclusion, heterozygous loss-of-function variants in SON define a recognizable syndrome, ZTTK, associated with a broad, severe phenotypic spectrum, characterized by a large inter-individual variability. These observations provide essential information for affected individuals, parents, and healthcare professionals to ensure appropriate clinical management.
Similar content being viewed by others

Introduction
In recent years, an increasing number of genetic causes of intellectual disability (ID) and developmental disorders have been discovered [1,2,3]. Heterozygous loss-of-function variants in SON (OMIM #182465) were first described in 2016 as one of these novel genetic causes [4,5,6,7] and the associated syndrome was subsequently named the Zhu–Tokita–Takenouchi–Kim syndrome (ZTTK syndrome, OMIM #617140). SON, located on 21q22.11, is a highly conserved gene that is ubiquitously expressed, with the most prominent isoform consisting of 12 exons (NM_138927.2 [8]). SON plays an important role in cell cycle progression and affects RNA splicing as a splicing cofactor [9, 10]. Haploinsufficiency of this gene can lead to intron retention and exon skipping, especially at weak splice sites, which affects multiple genes, including various genes involved in brain development, renal development, and metabolism [6, 8, 9, 11]. SON is involved in pluripotency and survival of embryonic stem cells as well as in the alternative splicing of other genes involved in epigenetic regulation and apoptosis [6, 8, 12]. Knockdown of SON in HeLa cells leads to several significant abnormal structures in cells that go through mitosis and on occasion even to mitotic arrest. Some of these abnormalities, like the protrusion of nuclear buds and micronuclei, implicate the role of SON in genomic stability [9]. Moreover, studies have shown that SON is involved in transcriptional regulation of, among others, leukaemia-associated genes and in tumorigenesis and haploinsufficiency of SON can cause neuronal migration defects and dendritic spine abnormalities [8, 13,14,15]. Taking into account the many different functions of SON, it is to be expected that pathogenic variants in this gene can cause diverse clinical symptoms.
It is therefore not surprising that ZTTK syndrome affects multiple organ systems and thus can induce a wide variety of symptoms. Reported clinical characteristics include ID/developmental delay, brain malformations, facial dysmorphisms, eye and/or vision abnormalities, heart defects, urogenital and gastrointestinal malformations, musculoskeletal abnormalities, short stature, and craniosynostosis [4,5,6,7]. Knockdown of SON in zebrafish results in a phenotype with morphological brain abnormalities for instance, which resembles (part of) the characteristics that are described in individuals with the ZTTK syndrome [6].
Although pathogenic variants in SON have been described in multiple publications [4,5,6,7, 11, 16,17,18,19,20], a systematic review was not yet available and an accurate genotype–phenotype study has not yet been performed. Here, we review the phenotype of 52 individuals with variants in SON, including 17 previously unpublished individuals, to establish the phenotypic spectrum associated with ZTTK syndrome. In addition, we assessed the facial gestalt and compared the phenotype of individuals with de novo SON loss-of-function variants and those with a de novo SON missense variant. This knowledge is essential for both professionals, as well as for affected individuals and their families, as it may guide clinical management and provide insights for accurate counseling and prognosis.
Methods
Identification of individuals with a variant in SON
A literature search was conducted in PubMed to collect all published data on clinical consequences of a pathogenic variant in SON. The following search terms were used in August of 2020:
(“SON protein, human” [Supplementary Concept] OR SON[ti] OR SON3[ti] OR BASS1[ti] OR DBP-5[ti] OR NREBP[ti] OR TOKIMS[ti] OR C21orf50[ti] OR SON[ot] OR SON3[ot] OR BASS1[ot] OR DBP-5[ot] OR NREBP[ot] OR TOKIMS[ot] OR C21orf50[ot]) AND (“Mutation”[Mesh] OR “Genome”[Mesh] OR mutat*[Title/Abstract] OR variant*[tiab]) OR Zhu-Tokita-Takenouchi-Kim[tiab] OR ZTTK[tiab]
In addition, Gene PubMed was searched for SON and all linked articles were checked for the availability of phenotypic description of individuals. Articles were selected based on title and abstract. Inclusion criteria were: a clinically relevant variant in SON, availability of clinical data and availability of full text in English or Dutch. After inclusion, the full texts were assessed. Articles were excluded after full text analysis if the clinical characteristics or the exact variant were not described, or if the variant or phenotype was not trackable to a specific individual. The references of the included articles were manually assessed for other relevant articles. Two researchers (AJMD and KMGT) independently performed the search and selection of articles to ensure that all articles meeting the criteria were included.
Furthermore, an additional cohort of 17 previously unpublished individuals was recruited through gene-matcher exchange programs and direct intercollegiate contacts (all data in Supplementary Table 1). For two individuals (16 and 52), the information from the initial publication was updated and completed here.
Genetic and phenotypic data
Data on genomic and phenotypic abnormalities from previously published cases were extracted from tables, figures, text, and supplements of the published articles. All other genomic variants potentially contributing to the phenotype of the patients as listed in the original publications are provided in Supplementary Table 2. A potential contribution of these variant to the phenotype cannot be excluded. All other genomic variants potentially contributing to the phenotype of the patients as listed in the original publications are provided in Supplementary Table 2. A potential contribution of these variant to the phenotype cannot be excluded. For the novel cases, clinicians were asked to supply clinical and genomic data. To create an accessible and adaptable overview, all phenotypic data were collected in Human Phenotype Ontology (HPO) [21] terms, and subsequently entered in the SON page of the “Human Disease Genes (HDG)“ website series [22] allowing the display of graphic representations of the clinical data. The frequency of each abnormality was determined to get a complete overview of the phenotypic spectrum. A separate overview was made for the observed phenotypic abnormalities in individuals with a missense variant in SON. St. Jude’s ProteinPaint was used to visualize the genomic variants in SON [23]. Finally, all genetic variants of previously unpublished individuals were uploaded to the LOVD (https://databases.lovd.nl/shared/individuals/SON, ID #00377114 and #00377116-#00377132) to ensure availability of genotypic and phenotypic data.
Functional validation
Functional analysis of missense variants was performed when patient material was available, and as described previously by Kim et al. [6]. In brief, SON haploinsufficiency has previously been shown to cause erroneous splicing of targeted genes, which can be visualized by intron retention and/or exon skipping using PCR at cDNA level, as well as reduced gene expression levels by qPCR as a consequence of nonsense-mediated decay of the miss-spliced mRNA products. To functionally assess the missense variant observed in individual 18, intron retention/exon skipping for ACY1, PNPK, TUBG1, WDR62, PFKL, and PSMD3 was tested, as well as the expression levels of ADA and HDAC6.
Quantitative facial phenotyping
Quantitative facial phenotyping was used to analyze the individual’s facial morphology. Photographs of 26 individuals were analyzed using the Hybrid model, as described previously [24,25,26]. With this model, the clustering impact factor (CIF) of individuals with a variant in SON can be determined. The CIF reflects the extent to which these individuals cluster within a group of controls and thus to which extend their facial features are similar. The controls are age-, gender-, and ethnicity-matched individuals with ID. To determine whether the CIF was significantly higher than can be expected by random chance, the Mann–Whitney U test was used.
Results
Cohort
The PubMed search yielded 343 articles. After evaluation based on in- and exclusion criteria, 9 articles remained (Supplementary Fig. 1), in which data on 35 individuals with presumed clinically relevant SON variants were published [4,5,6,7, 11, 16,17,18, 20]. Manual assessment of the references did not lead to inclusion of additional articles. No discrepancies were identified between the two researchers who performed the search and selection.
Through international collaboration, 17 unpublished individuals with presumed pathogenic variants in SON were identified and clinically characterized (Supplementary Table 1), resulting in a total cohort size of 52 individuals.
Genotypic variants
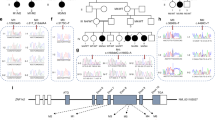
Detailed annotations for all variants identified in the 52 individuals identified after systematic review are provided in Supplementary Table 2, including variant annotations at genomic level (NC_000021.8), as well as their predicted protein effects based on NM_138927.2. In these 52 individuals, 40 frameshift variants, 5 nonsense variants, 2 in-frame deletions, 3 missense variants, and 2 whole gene deletions were observed. Of these variants, 49 were de novo and for 3 variants, the inheritance pattern could not be determined. Five variants were reported in multiple individuals: a 4-bp deletion, NC000021.8:g.34927290_34927293del, predicted to lead to p.(Val1918Glufs*87), was found in 13 individuals; three other small deletions, NC000021.8:g.34923418_34923419del (p.(Val629Alafs*56)), NC000021.8:g.34925389_34925393del (p.(Met1284Ilefs*2)), and NC000021.8:g.34927547del (p.(Val2004Trpfs*2)), as well as one substitution, NC000021.8: g.34924871C>T (p.(Arg1112*)), were each identified in two individuals. All variants were found in exon 3 and spread out evenly across this exon, except for one, NC000021.8:g.34929534del (p.(Pro2078Hisfs*4)), in exon 4 (Fig. 1).

Annotation is based on NM_138927.2. Although one variant p.(Val1918Glufs*87) is frequently present in our study cohort, overall, the variants do not seem to cluster and are spread out over the gene. Larger whole gene deletions observed in two individuals are not shown.
Phenotype
Of 52 individuals, 26 (50%) were female. The median age at examination was 6 years and 8 months (range: 1 year and 9 months to 34 years). On average, 21.2 HPO terms were assigned per individual. Variation in the phenotype was extensive and abnormalities were described in all organ systems (Table 1 and Supplementary Table 2).
Individuals with a loss-of-function variant
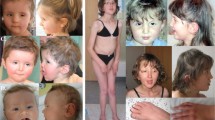
All but 1 of the 49 individuals with a loss-of-function variant had ID. The severity of the ID varied from mild (21%), to moderate (29%), to severe (50%). In 27 of 49 individuals (55%), an abnormality of prenatal development or birth was reported: multiple individuals were born premature (13/49, 27%) and/or with a birthweight below the third centile (12/36, 33%). A short stature (26/48, 54%), small head circumference (11/40, 28%), and low weight (14/25, 56%) were common symptoms. None of the individuals reported a growth parameter above the 97th percentile. In 44 of 48 individuals (92%), neurological abnormalities were described, in particular hypotonia (29/48, 60%), seizures (24/48, 50%), and an EEG abnormality (11/48, 23%). Brain imaging was performed in 41 of the individuals of whom 37 (90%) had an abnormality of the brain, with ventriculomegaly (24/41, 59%) being the most common abnormality. Also frequently observed were cortical dysplasia (7/41, 17%) and abnormalities of respectively the corpus callosum (18/41, 44%)—in particular hypoplasia of the corpus callosum (7/41, 17%), the cerebral white matter (6/41, 15%), and the cerebellum (6/41, 15%). Behavioral problems were mentioned in 26 out of 46 individuals (57%), with sleep disturbance (12/46, 26%) and autism (6/46, 13%) being particularly common. In 42 of 44 individuals (95%) for whom facial morphology was described, dysmorphisms were present (Fig. 2). Several dysmorphisms were reported recurrently, including facial asymmetry (13/44, 30%), horizontal eyebrows (14/44, 32%), downward slanting palpebral fissures (23/44, 52%), strabismus (21/44, 48%), deeply set eyes (12/44, 27%), epicanthal folds (9/44, 20%), abnormality of the nasal bridge (22/39, 56%), low-set ears (21/40, 53%), posteriorly rotated ears (6/40, 15%), midface retrusion (15/44, 34%), short or smooth philtrum (21/39, 54%), and thin upper lip vermillion (5/39, 13%). Musculoskeletal abnormalities occur frequently, in 40 out of 48 individuals (83%), including hypermobility (17/48, 35%) and pes planus (11/48, 23%). Furthermore, various other abnormalities of the feet (19/48, 40%), fingers (11/48, 23%), skull (10/48, 21%), and curvature of the vertebral column (7/48, 15%) were reported. Gastrointestinal abnormalities were seen in 32 of 45 individuals (71%), whereof feeding difficulties (29/45, 64%) and various structural abnormalities (10/45, 22%), especially those affecting the intestines, stomach, and gallbladder, were most common. In 15 out of 46 individuals (33%) cardiovascular abnormalities were observed, especially atrial septal defects (6/35, 17%) and ventricular septal defects (4/46, 9%). Abnormalities of the urogenital system were reported in 21 of 45 individuals (47%) with 17 individuals (38%) having an abnormality of the renal morphology, of whom 6 (13%) presented with a horseshoe kidney, and 6 of 45 (13%) had renal cysts. Of 46 individuals, 19 (41%) had an abnormality of the skin, hair or nails and 8 of 46 individuals (17%) had an abnormality of the teeth, mostly affecting the dental enamel. Also, visual impairments were observed in multiple individuals (21/47, 45%), especially hypermetropia (9/47, 19%) and cortical visual impairment (5/47, 11%). A hearing impairment was reported in 7 individuals (out of 46, 15%). An abnormality of the immune system was described in 15 of 46 individuals (33%), which among others leads to recurrent otitis media (6/46, 13%). Several abnormalities of the metabolic system have been described (10/46, 22%) and abnormalities of the endocrine system occurred in 6 of 43 individuals (14%). Neoplasia was not described in any of the individuals.

Adult individuals with SON haploinsufficiency
Our cohort includes five individuals who had reached adult age at the time of examination: individuals 13, 34, 37, 41, and 50. To see whether any symptoms were more prominent at an older age—which might indicate that these problems develop or progress during life—we looked at this subgroup with special interest (Supplementary Table 2).
Similar to the younger individuals, all adults had ID (5/5, 100%). Three (60%) were reported to have severe ID, while mild and moderate ID were both seen once (20%). Hypotonia was present in three (60%) adults. Brain imaging reported some abnormality of the brain (4/4, 100%) in all adult cases, mostly ventriculomegaly (3/4, 75%). Three out of five (60%) adults were diagnosed with behavioral problems, of whom two with autism (40%). Symptoms of the musculoskeletal system were identified in all adults (5/5, 100%), with contractures (2/4, 50%) and abnormalities of the curvature of the vertebral column (3/4, 75%) being prominently present. Of note, 2/3 individuals with a curvature of the vertebral columns had hypotonia as well, indicating it might be secondary to the lower muscle tone. In two individuals (2/4, 50%) hypertension was reported.
Individuals with a missense variant
We identified three individuals (one male, two females) with a unique heterozygous missense variant in SON (Fig. 1). In two, the variant occurred de novo whereas inheritance could not be determined for the last individual. SIFT predicted a deleterious effect for all three variants (score 0 for all), while MutationTaster predicted disease causing for p.(Ser1843Tyr) with probability 1 and polymorphism for the other two variants (probability 0.728 for p.(Thr579Ser) and 0.933 for p.(Ser223Leu)).Preliminary functional analysis of one of the de novo missense variants, p.(Thr579Ser), observed in individual 18, does not show the typical observed dysregulation of splicing of downstream targets (Fig. 3) as is observed for loss-of-function variants [6, 11]. Patient material for functional analysis of the other two missense variants was not available. Looking at the phenotype of individuals with a missense variant in SON, one individual had severe ID, while one did not have ID. For the third individual, ID was not reported, but motor delay was. One individual was born premature. In two individuals, hypotonia was observed. Abnormalities of the brain were absent in all three individuals after brain imaging. Aggressive behavior was seen in one individual. All three had facial dysmorphisms, including abnormalities of the eye (consisting of abnormal eye movement and ptosis), and/or epicanthal folds, and abnormalities of the nose, mouth, and ears in two. Musculoskeletal abnormalities were described in two of the individuals, as were abnormalities of the skin, hair, and/or nails. One individual reported gastrointestinal abnormalities, among which feeding difficulties. In another individual, a ventricular septal defect was seen. In one individual, osteochondroma and haemangiomas were observed. No abnormalities of the renal morphology, vision, hearing, immunological system, endocrine system, or metabolic system were observed.

A Real-time qPCR detecting the levels of SON and SON-targeted transcripts from Individual 18 with a SON missense variant (p.(Thr579Ser)). Three different pairs of the primers were used to measure the level of SON mRNA. Overall, the levels of SON and previously known targets of SON-mediated RNA splicing were not significantly downregulated in the affected individual. The mRNA levels of indicated genes were normalized to the level of TUBA1A mRNA. Errors bars represent mean ± SD from three technical replicates. For LoF variants, previous work [6] showed reduced expression of all genes tested in panel A, as well as aberrant splicing of exons as indicated in panel B. B In the reverse transcription and PCR analysis, exon skipping events in ADA and HDAC6, which were previously confirmed in individuals with SON loss-of-function variants, were not identified in Individual 18 with a SON missense variant (p.(Thr579Ser)).
Quantitative facial phenotyping
Quantitative facial phenotyping was used to analyze the facial morphology in 26 photos, including 25 individuals with de novo loss-of-function variants and one with a de novo missense variant. No significant clustering was found between individuals (CIF = 0.87461, p = 0.37), indicating the hybrid model could not detect a specific recognizable facial phenotype.
Discussion
This study presents an up-to-date overview of the phenotypic spectrum of 52 individuals with a variant in SON, including 17 novel, previously unpublished, individuals. In 49 individuals, the variants are predicted to lead to haploinsufficiency, whereas we also present data of three individuals with missense variants of which the underlying pathophysiological mechanism seems different.
Phenotypic spectrum caused by SON haploinsufficiency
A broad variety of phenotypic abnormalities was observed in the individuals with a loss-of-function variant in SON (Supplementary Table 2) and multiple organ systems were affected. This is in line with the function of SON as it affects, among others, RNA splicing and transcription of multiple genes [6, 8]. Several of these downstream targets are known to lead to Mendelian phenotypes when disrupted, each of which is also observed in individuals with SON haploinsufficiency [6,7,8].
The variety of symptoms caused by SON haploinsufficiency reported previously is also observed in our study. Moreover, our study is now able to substantiate the suggested association of several specific symptoms to this syndrome. Whereas neurological, musculoskeletal, developmental, visual, and congenital abnormalities have been reported recurrently before, one of the new associations was previously made by Quintana Castanedo et al. who stated in a case report that abnormalities of the nails might be a feature of ZTTK syndrome that had not been published before [16]. Abnormalities of the skin, hair, or nails were described in 18 other individuals included in this study (41%), of whom 5 (11%) had an abnormality of the nails. Our findings therefore confirm that such abnormalities are part of ZTTK syndrome. Renal abnormalities have also been suggested as an important feature of ZTTK syndrome [11], and were collectively reported now in 21 individuals (47%). Despite growth hormone deficiency not being mentioned as a feature of ZTTK syndrome in previous studies, it was present in three individuals. In addition, in three other individuals effective treatment of short stature with growth hormone was initiated.
Using quantitative facial phenotyping no clustering was found in individuals with a variant in SON compared to controls. This means that although several facial dysmorphisms were described recurrently, our algorithm did not detect a clear recognizable facial phenotype underscoring the clinical observation that there is no consistent characteristic facial gestalt. An à vue clinical diagnosis of ZTTK syndrome based on facial abnormalities should therefore always be supported by whole exome sequencing (WES) or targeted gene analysis.
The inclusion of previously published individuals has some restrictions as available data depend on how the author has interpreted and described the phenotypic abnormalities, and in some articles, not all organ systems were described. For missing information, the completeness of the article was used to determine the probability of data being not reported or not present in the individual, which may have led to misinterpretation. However, by setting up the study this way, a significant amount of data could be used and therefore a complete, up-to-date, overview could be provided.
Adult individuals
Most symptoms (like ID and the severity of ID) were not reported more frequently in the five adult individuals compared to the individuals under the age of 18. However, while only three individuals (3/41, 7%) with a truncating variant in SON younger than 18 years old were diagnosed with autism, 50% (2/4) of adults had autism. As for hypertension, 50% (2/4) of adult individuals reportedly had this from a relatively young age (23 and 34 years), indicating a possible risk on early onset hypertension.
SON missense variants
In the three individuals with a missense variant, diverse phenotypic abnormalities were observed as well. The variants in these three individuals remain variants of unknown clinical significance. At the clinical level, the only notable difference in phenotype between individuals with a missense and a truncating variant is that in none of the individuals with a missense variant a brain abnormality was noted, while these were present in 90% of individuals with a truncating variant. For the other features, the phenotype of these individuals was in line/overlapping with those having loss-of-function variants.
At functional, however, the missense variant tested does not show the typical dysregulation of splicing or downregulation of genes regulated by SON as was previously established. For SON haploinsufficiency, splice defects are seen for genes involved in neuronal migration, cortex organization, and metabolism as well as genes involved in congenital abnormalities of the kidney and urinary tract. As for instance, these neuronal migration defects are not observed in individuals with de novo missense variants, these data may suggest that de novo missense variants cause a phenotype overlapping with ZTTK, but are caused via different, yet to be established, pathophysiological mechanisms. In combination with population constraint metrics from The Genome Aggregation Database (gnomAD, v.2.1.1), reporting a pLI of 1.0 for loss-of-function variants, and a Z-score of 1.54 for missense variants, our data may also suggest that the SON de novo missense variants are not related to the phenotype observed, and that the phenotype in these individuals is caused by a yet to be identified other genetic factor. Analysis of the WES data of each individual with a SON missense variant has, however, failed to identify another such variant.
Along the same rationale, there is also a report of a 3-year old boy, born to consanguineous parents, with a homozygous missense variant in SON (p.(Ala557Val)) [19]. He was diagnosed with a medulloblastoma and café-au-lait spots. Both parents were reported to have the same skin lesions, but no history of neoplasia. In the absence of detailed phenotypic and further genotype information, this report does not provide further insights in establishing the role of missense variants in ZTTK, or another overlapping, syndrome.
Clinical recommendations
Based on the phenotypic abnormalities described in this study, we provide the following suggestions for follow-up of individuals with ZTTK syndrome, caused by SON haploinsufficiency. We recommend neurological screening with special attention for hypotonia, seizures, and morphological abnormalities of the brain (Table 2). In addition, because of its common occurrence, feeding difficulties should be monitored. We also recommend cardiac and renal screening, as well as screening for visual impairment. Lastly, routine repeated monitoring of the individuals is recommended—especially for autism and hypertension, since these might develop at a later age—because of the wide range of symptoms described and the uncertainty of the natural course of this syndrome.
As this syndrome is rare, and has not yet been fully elucidated, it is necessary to keep collecting information on phenotypic abnormalities. A dedicated ZTTK syndrome website to collect such information as part of the HDG Website series is available at www.humandiseasegenes.info/son. To ensure the most up-to-date information for genetic counselors and individuals and their families, additional cases can still be added—even after publication of this current study.
Conclusion
In conclusion, this study provides a systematic phenotypic overview of 52 individuals with a (likely) pathogenic variant in SON. A broad spectrum of abnormalities was observed, affecting multiple organ systems, with a high clinical inter-individual variability. We confirm that heterozygous loss-of-function variants in SON define the clinically recognizable ZTTK syndrome.
This overview is of great value for affected individuals, parents, and healthcare professionals as it provides more insight into the natural course of disease and provides suggestions for how individuals should be followed up and clinically managed.
Data availability
The data that support the findings of this study are available in the Supplementary material of this article and online at https://databases.lovd.nl/shared/genes/SON and https://humandiseasegenes.nl/son/.
References
de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–9.
Vissers LE, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, et al. A de novo paradigm for mental retardation. Nat Genet. 2010;42:1109–12.
Vissers LE, Gilissen C, Veltman JA. Genetic studies in intellectual disability and related disorders. Nat Rev Genet. 2016;17:9–18.
Zhu X, Petrovski S, Xie P, Ruzzo EK, Lu YF, McSweeney KM, et al. Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet Med. 2015;17:774–81.
Tokita MJ, Braxton AA, Shao Y, Lewis AM, Vincent M, Küry S, et al. De novo truncating variants in SON cause intellectual disability, congenital malformations, and failure to thrive. Am J Hum Genet. 2016;99:720–7.
Kim JH, Shinde DN, Reijnders MRF, Hauser NS, Belmonte RL, Wilson GR, et al. De novo mutations in SON disrupt RNA splicing of genes essential for brain development and metabolism, causing an intellectual-disability syndrome. Am J Hum Genet. 2016;99:711–9.
Takenouchi T, Miura K, Uehara T, Mizuno S, Kosaki K. Establishing SON in 21q22.11 as a cause a new syndromic form of intellectual disability: possible contribution to Braddock-Carey syndrome phenotype. Am J Med Genet A. 2016;170:2587–90.
Hickey CJ, Kim JH, Ahn EY. New discoveries of old SON: a link between RNA splicing and cancer. J Cell Biochem. 2014;115:224–31.
Ahn EY, DeKelver RC, Lo MC, Nguyen TA, Matsuura S, Boyapati A, et al. SON controls cell-cycle progression by coordinated regulation of RNA splicing. Mol Cell. 2011;42:185–98.
Sharma A, Markey M, Torres-Muñoz K, Varia S, Kadakia M, Bubulya A, et al. Son maintains accurate splicing for a subset of human pre-mRNAs. J Cell Sci. 2011;124:4286–98.
Kim JH, Park EY, Chitayat D, Stachura DL, Schaper J, Lindstrom K, et al. SON haploinsufficiency causes impaired pre-mRNA splicing of CAKUT genes and heterogeneous renal phenotypes. Kidney Int. 2019;95:1494–504.
Lu X, Göke J, Sachs F, Jacques P, Liang H, Feng B, et al. SON connects the splicing-regulatory network with pluripotency in human embryonic stem cells. Nat Cell Biol. 2013;15:1141–52.
Ueda M, Matsuki T, Fukada M, Eda S, Toya A, Iio A, et al. Knockdown of Son, a mouse homologue of the ZTTK syndrome gene, causes neuronal migration defects and dendritic spine abnormalities. Mol Brain. 2020;13:80.
Kim JH, Baddoo MC, Park EY, Stone JK, Park H, Butler TW, et al. SON and its alternatively spliced isoforms control MLL complex-mediated H3K4me3 and transcription of leukemia-associated genes. Mol Cell. 2016;61:859–73.
Ahn EE, Higashi T, Yan M, Matsuura S, Hickey CJ, Lo MC, et al. SON protein regulates GATA-2 through transcriptional control of the microRNA 23a~27a~24-2 cluster. J Biol Chem. 2013;288:5381–8.
Quintana Castanedo L, Sánchez Orta A, Maseda Pedrero R, Santos Simarro F, Palomares Bralo M, Feito Rodríguez M, et al. Skin and nails abnormalities in a patient with ZTTK syndrome and a de novo mutation in SON. Pediatr Dermatol. 2020;37:517–9.
Tan Y, Duan L, Yang K, Liu Q, Wang J, Dong Z, et al. A novel frameshift variant in SON causes Zhu-Tokita-Takenouchi-Kim Syndrome. J Clin Lab Anal. 2020;34:e23326.
Yang Y, Xu L, Yu Z, Huang H, Yang L. Clinical and genetic analysis of ZTTK syndrome caused by SON heterozygous mutation c.394C>T. Mol Genet Genom Med. 2019;7:e953.
Chiu C, Loth S, Kuhlen M, Ginzel S, Schaper J, Rosenbaum T, et al. Mutated SON putatively causes a cancer syndrome comprising high-risk medulloblastoma combined with café-au-lait spots. Fam Cancer. 2019;18:353–8.
Slezak R, Smigiel R, Rydzanicz M, Pollak A, Kosinska J, Stawinski P, et al. Phenotypic expansion in Zhu-Tokita-Takenouchi-Kim syndrome caused by de novo variants in the SON gene. Mol Genet Genom Med. 2020;8:e1432.
Robinson PN, Köhler S, Bauer S, Seelow D, Horn D, Mundlos S. The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. Am J Hum Genet. 2008;83:610–5.
Dingemans AJM, Stremmelaar DE, Vissers L, Jansen S, Nabais Sá MJ, van Remortele A, et al. Human Disease Genes website series: an international, open and dynamic library for up-to-date clinical information. Am J Med Genet A. 2021;185:1039–46.
Portal SJCsRHPD. ProteinPaint. 2015.
van der Donk R, Jansen S, Schuurs-Hoeijmakers JHM, Koolen DA, Goltstein L, Hoischen A, et al. Next-generation phenotyping using computer vision algorithms in rare genomic neurodevelopmental disorders. Genet Med. 2019;21:1719–25.
Dingemans AJM, Stremmelaar DE, van der Donk R, Vissers L, Koolen DA, Rump P, et al. Quantitative facial phenotyping for Koolen-de Vries and 22q11.2 deletion syndrome. Eur J Hum Genet. 2021. [epub ahead of print]
Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6:e1000097.
Acknowledgements
First and foremost, we are grateful to all individuals and their parents for participation in this study. Next to that, we are grateful to the Dutch Organisation for Health Research and Development: ZON-MW grants 912-12-109 (to BBAdV and LELMV), Donders Junior researcher grant 2019 (to BBAdV and LELMV) and Aspasia grant 015.014.066 (to LELMV). This study was also supported by the US National Institute of Health grants R01 CA190688 and R01 CA236911 (to E-YEA). This work has been generated through a collaboration with the European Reference Network on Rare Congenital Malformations and Rare Intellectual Disability (ERN-ITHACA) [EU Framework Partnership Agreement ID: 3HP-HP-FPA ERN-01-2016/739516]. The aims of this study contribute to the Solve-RD project (to LELMV) that has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 779257.
Author information
Authors and Affiliations
Contributions
Conceptualization: A.J.M.D, K.M.G.T, E.Y.E.A., B.B.A.d.V, L.E.L.M.V; Data collection: all authors; Data Analysis: A.J.M.D, K.M.G.T; Funding acquisition: L.E.L.M.V, B.B.A.d.V, E.Y.E.A.; Writing – original draft: A.J.M.D, K.M.G.T, E.Y.E.A., B.B.A.d.V, L.E.L.M.V; Writing – review and editing: all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Consent for participation in this study was obtained via the individuals’ legal guardians, and the study was approved by the institutional review board of the Radboud University Medical Center (#2020–6763). For individuals whose facial photos are depicted in Fig. 2, additional informed signed consent was obtained for the publication of photographs.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Dingemans, A.J.M., Truijen, K.M.G., Kim, JH. et al. Establishing the phenotypic spectrum of ZTTK syndrome by analysis of 52 individuals with variants in SON. Eur J Hum Genet 30, 271–281 (2022). https://doi.org/10.1038/s41431-021-00960-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-021-00960-4
This article is cited by
-
Recurrent myocardial injury in a de novo SON mutation ZTTK syndrome patient: a case report
BMC Pediatrics (2024)
-
Istore: a project on innovative statistical methodologies to improve rare diseases clinical trials in limited populations
Orphanet Journal of Rare Diseases (2024)
-
Nuclear speckleopathies: developmental disorders caused by variants in genes encoding nuclear speckle proteins
Human Genetics (2023)
-
Good genotype-phenotype relationships in rare disease are hard to find
European Journal of Human Genetics (2022)
-
Commentary on: Establishing the phenotypic spectrum of ZTTK syndrome by analysis of 52 individuals with variants in SON
European Journal of Human Genetics (2022)
