
Abstract
Background/objectives
Thyroid-associated ophthalmopathy (TAO), an autoimmune component of Graves’ disease, remains a disfiguring and potentially blinding condition. Here, the author reviews the role of insulin-like growth factor-I receptor pathway in TAO and how it might be therapeutically targeted.
Methods
The recent literature is reviewed.
Results
TAO involves reactivity of orbital connective tissues and their remodeling. While many of the details concerning the pathogenesis of TAO remain to be determined, several insights have come to light recently. Among them is the apparent involvement of IGF-IR. This receptor protein, a membrane-spanning tyrosine kinase receptor can form both physical and functional complexes with the thyrotropin receptor (TSHR). This is notable because TSHR is the established primary autoantigen in Graves’ disease. IGF-IR activity is critical to signaling downstream from both IGF-IR and TSHR. In addition, antibodies against IGF-IR have been detected in patients with Graves’ disease and in rodent models of TAO. Evidence has been put forward that these antibodies may act directly on IGF-IR, perhaps in some manner activating the receptor. These experimental observations have led to the development of a novel therapy for active TAO, utilizing a monoclonal anti-IGF-IR inhibitory antibody which had been produced originally as treatment for cancer. The agent, teprotumumab was recently evaluated in a clinical trial and found to be highly effective and relatively well-tolerated. It is currently undergoing assessment in a follow-up trial.
Conclusions
Should the current study yield similarly encouraging results, it is possible that teprotumumab will emerge as a paradigm-shifting medical therapy for TAO.
Similar content being viewed by others
Introduction to the insulin-like growth factor-I receptor
The insulin-like growth factor-I (IGF-I) pathway plays critical roles in the regulation of cell metabolism, survival, and growth [1, 2]. The pathway comprises both IGF-I and IGF-II, two surface receptors, including IGF-I receptor (IGF-IR) and IGF-IIR/mannose-6-phosphate receptor, six IGF-I binding proteins and nine IGF-I binding protein-related proteins [2,3,4]. Its involvement in immune function has been recognized for several decades and is now being considered as a target for therapy in human autoimmune diseases [5]. IGF-IR is a membrane-spanning tyrosine kinase protein that can bind IGF-I and IGF-II [6]. It can also be activated by insulin although IGF-I is its preferred agonist ligand. It exhibits a heterotetrameric structure that includes an extracellular ligand binding domain located in two α subunits and a kinase domain located in two β subunits. These subunits are linked by two disulphide bonds. Further, IGF-IR and the insulin receptor can form heterodimers and many tissues, such as fat, may be dominated by hybrid receptors [7, 8]. Human IGF-IR is encoded by a gene located on chromosome 15. The receptor is ubiquitously expressed in many tissues and cell types. Its activities are regulated by several proteins, among them the IGF-I binding proteins which govern the interactions between IGF-IR and activating ligands [3]. Substantial evidence supports the concept that IGF-IR participates in the pathogenesis of several forms of cancer [9]. This realization resulted in the initiation of several drug development programs at multiple pharmaceutical companies [10]. Most of these programs have been terminated because these drugs failed to exhibit encouraging effectiveness against several forms of cancer. Recent insights into the signaling downstream from IGF-IR have added several layers of complexity to how we now view the central importance of this pathway in human physiology and disease [11].
Evidence for IGF-IR involvement in Graves’ disease
Graves’ disease (GD) represents an autoimmune syndrome involving the thyroid, orbital connective tissues, and specific regions of the skin [12]. The central autoantigen in GD is the thyrotropin receptor (TSHR). Activating antibodies directed against TSHR, known as thyroid-stimulating immunoglobulins (TSI), are directly responsible for the hyperthyroidism frequently occurring in GD [13]. The role of TSHR and TSI in the development of thyroid-associated ophthalmopathy (TAO) remains less well defined although substantial evidence, much of it circumstantial, supports their involvement. Emerging insights suggest that a second cell surface receptor might also play an important role in GD and in TAO [14]. A major obstacle to better defining the underlying pathogenesis of TAO has been the historical absence of a high-fidelity animal model for the disease although recent progress in developing these models now offers a promising platform for preclinical investigation [15]. The first clue that IGF-IR might be involved in TAO was provided by Weightman and colleagues [16]. They had speculated that earlier observations concerning immunoglobulins from patients with GD (GD-IgG) stimulating fibroblasts and extraocular myoblasts [17, 18] might be acting through IGF-IR. They reported that IgG collected from patients with GD, regardless of whether or not they manifested TAO, could displace radiolabeled IGF-I from binding sites on the surfaces of orbital fibroblasts generated from tissues explanted from extraocular muscle. Those binding sites had an apparent Kd of 0.5 nM. In contrast, IgG from healthy controls failed to alter IGF-I binding to these cells. Independently, Ingbar and colleagues laid the groundwork for many of the studies that have followed dissecting the relationship between the thyroid axis and growth hormone/IGF-I pathways. They had previously found that IGF-I and insulin could synergistically enhance the actions of TSH and TSI on thyroid epithelial cells in culture [19].
More recently, Pritchard et al. [20] demonstrated that IGF-IR is overexpressed by orbital fibroblasts from patients with TAO. Interrupting IGF-IR activity, either with inhibitory anti-IGF-IR antibodies or by transfecting orbital fibroblasts with a dominant negative receptor, could abolish signaling initiated by GD-IgG [20]. The signaling mediating the actions of GD-IgG in these cells has been mapped to the FRAP/mTor/p70sk6 pathway and is rapamycin-sensitive [21]. Subsequently, T cells were also found to overexpress IGF-IR [22] and the relative abundance of IGF-IR-displaying B cells in GD [23] was found to be increased. Thus, several clues existed that IGF-IR might be involved in GD and TAO, providing the rationale for considering this pathway as a potential therapeutic target.
Evidence that antibodies against IGF-IR might be generated in GD
The studies from Weightman et al [16] suggested that GD-IgG might include molecules exhibiting affinity toward a fibroblast surface-displayed protein that also binds IGF-I. While those studies identified IGF-IR as a strong candidate for this binding, Pritchard et al. [20] ultimately established the identity of the binding site as IGF-IR. Using the specific IGF-I analogue, Des 1-3, they showed that the principal IGF-I binding site on orbital fibroblasts is IGF-IR and that GD-IgG could compete for this site. Their studies further demonstrated that GD-IgG could mimic the actions of IGF-I while rhTSH failed to do so [20]. Among its effects, GD-IgG was shown to induce the expression of the chemokine known as “regulated on activation normal T cell expressed and secreted” (RANTES) and IL-16 [20, 21]. These two T cell chemoattractants play important roles in the trafficking of cells to sites of tissue damage and remodeling [24, 25]. The existence of antibodies directed at IGF-IR, and especially those with IGF-IR-activating properties, as distinct from anti-TSHR antibodies, has been contentious. Some studies have failed to detect these [26, 27] while others have demonstrated anti-IGF-IR antibodies [16, 28, 29]. Further, anti-IGF-IR antibodies have been detected in a mouse model of GD manifesting some features of TAO [30]. At the heart of the debate is whether all activities of GD-IgG can correctly be attributed to those antibodies activating TSHR. Speaking against that possibility are the findings of Pritchard et al. [31] demonstrating identical IGF-IR signal-initiating activities in sera and IgGs from patients with rheumatoid arthritis but not manifesting GD. Underlying the divergent findings thus far reported may be the substantially different assays used to detect anti-IGF-IR antibodies and lack of standardization of the cell models used in each case. In any event, the issue of whether anti-IGF-IR antibodies, generated as a consequence of disease, can in some manner directly initiate signaling through that receptor is yet to be settled (Fig. 1).
Evidence that IGF-IR and TSHR might interact
The initial evidence for an overlap between TSH and IGF-I (and insulin) pathways dates from the studies of Ingbar and colleagues 30 years ago [19]. They reported that both IGF-I and insulin could enhance the actions of TSH and TSIs in cultured FRTL5 cells. The relationship between IGF-IR and TSHR was demonstrated initially by Tsui et al. [32] who found that the two membrane-spanning proteins co-localize under confocal microscopy of orbital fibroblasts, cultured human thyroid epithelial cells, and in situ in orbital fat derived from patients with TAO. Their studies included co-immunoprecipitation studies which revealed that the two receptor proteins could be brought out of the solution with monoclonal antibodies against either receptor. With regard to co-dependent function, the study further demonstrated that an inhibitory monoclonal antibody directed against IGF-IR (1H7) could block signaling initiated at either IGF-IR or TSHR. Specifically, the activation of Erk by IGF-I, rhTSH, or IgGs from patients with GD could be attenuated by 1H7 [32]. In aggregate, that study strongly suggested the potential for interruption of IGF-IR activity as a therapy for diseases mediated by IGF-I, TSI, and activating antibodies directed against both receptors.
Organization of clinical trials based on findings in vitro
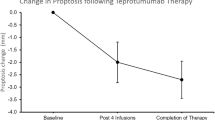
A multicenter randomized, placebo-controlled, double-masked clinical trial was conducted to determine whether inhibiting IGF-IR with a fully human, monoclonal antibody was effective and safe in moderate to severe, active TAO [33] (Fig. 2). Study enrollment was limited to patients who were 18–75 years old, who had manifested TAO within 9 months of study entry, who had clinical activity scores (CAS) of ≥4 points on a 7-point scale and whose eye disease was judged to be moderate to severe in the more severely affected (study) eye. The study design included baseline assessment, entry into a 24-week treatment period during which patients received 8 infusions at 3 weekly intervals followed by a 48-week follow-up. The primary response, assessed at week 24 (3 weeks after the final infusion), comprised the aggregate of a reduction of ≥2 points in CAS and a reduction of ≥2 mm in proptosis in the study eye without a similar magnitude of worsening in the fellow eye. Secondary endpoints included reduction in proptosis, improvement in CAS, improvement in the quality of life using a validated instrument, and improved diplopia as independent variables. A total of 112 patients underwent screening of which 88 were randomized to either the treatment arm receiving teprotumumab or placebo in a 1:1 ratio. Thirty-nine patients receiving placebo (87%) and 37 patients in the teprotumumab group (88%) completed the intervention.
Twenty-nine patients of forty-two (69%) receiving teprotumumab in the intention to treat cohort responded at 24 weeks as compared to 9 of 45 patients in the placebo group (20%) (p < 0.001). Patients receiving the drug had a greater reduction in proptosis (p < 0.001), CAS (p < 0.001), and improved quality of life at 24 weeks compared to placebo. Time to first response was substantially shorter in the teprotumumab group than in those receiving placebo. Eighteen of forty-two patients (43%) receiving active drug achieved the primary endpoint at 6 weeks, indicating a very rapid response to teprotumumab. In contrast, 2 of 45 patients (4%) receiving placebo responded at 6 weeks (p < 0.001). With regard to safety, hyperglycemia detected in patients with preexisting diabetes was the only drug-related adverse event and was easily controlled with adjustment of diabetes medication. This increased requirement in medication was transient and returned to baseline following the conclusion of exposure to teprotumumab.
Thus, in patients with active, moderate to severe TAO, inhibition of IGF-IR with teprotumumab was more effective than placebo in improving CAS and proptosis while exhibiting an encouraging safety profile. On the basis of the results of this phase II trial, the US FDA designated teprotumumab as a breakthrough therapy for TAO. Long-term follow-up results are now being analyzed. In addition, a second, confirmatory trial was organized and has now completed enrollment. That trial, like the first, is limited to patients with relatively recent onset of TAO (≤9 months) but has a simplified primary endpoint consisting of reduction in proptosis of ≥2 mm in the study eye with no similar magnitude worsening in the fellow eye. In addition, all non-responders, regardless of whether they were in the teprotumumab cohort or those who had received placebo during the treatment phase, are offered a 24-week treatment of the active drug as an open label study phase.
The reported trial has limitations. Despite attempts to stratify participants with tobacco smoking histories, an imbalance existed. The study did not include orbital imaging that would allow pre- and post-treatment comparisons. This could have helped determine whether teprotumumab targeted extraocular muscles, adipose/connective tissue, or both. The duration of follow-up observation has thus far been inadequate to fully reveal the durability and long-term safety of the drug. We anticipate that these gaps in our current understanding will be filled with subsequent investigations.
Implications of the findings from clinical trials of teprotumumab in TAO
The positive results thus far reported for teprotumumab are unprecedented with regard to effectiveness. They appear to be equivalent to the best results thus far reported for surgical orbital decompressions in TAO. Thus they are likely to alter the patterns of medical care for this vexing and disfiguring condition should the currently conducted trial reveal similar effectiveness and safety. It is possible that other autoimmune diseases might similarly benefit from inhibition of the IGF-IR [5]. For instance, an identical mechanism for the involvement of that receptor was reported by our group in rheumatoid arthritis more than 1 decade ago [31] and thus that disease might represent an ideal candidate for further clinical study.

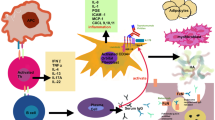
Theoretical model of the pathogenic events occurring in TAO. CD34+ fibrocytes of the monocyte lineage infiltrate the orbit. These cells express thyroid autoantigens in an autoimmune regulator protein-dependent manner. These proteins include thyrotropin receptor, thyroglobulin, thyroperoxidase, and iodide symporter. Fibrocytes present antigens to antigen-specific T cells which in turn support IgG1 production in B cells. Fibrocytes have the capacity to differentiate into CD34+ fibroblasts. Depending on the microenvironment, they can further differentiate into myofibroblasts and adipocytes. CD34+ fibroblasts co-populate the orbit with CD34− fibroblasts. When activated, fibrocytes and orbital fibroblasts produce several cytokines, including interleukins 1β, 6, 8, 10, 12, 16, TGF-β, tumor necrosis factor α, the chemokine known as “regulated on activation, normal T expressed and secreted” or RANTES, CXCL-12 and CD40 ligand (CD154). Virtually all infiltrating and residential orbital cells surface display insulin-like growth factor-I receptor (IGF-IR). Thus therapies based on the inhibition of IGF-IR might target any and all of these cells. Cytokine-activated orbital fibroblasts synthesize hyaluronan which can expand orbital connective tissue volume. Orbital fat can also expand from de novo adipogenesis. (From N Engl J Med, Smith TJ and Hegedus L, Graves’ disease, 375; 1552–1565. Copyright © (2016) Massachusetts Medical Society. Reprinted with permission)

Screening, randomization, response and follow-up of patients’ participation in a clinical trial of teprotumumab. a Patients entered the trial screening process after they had met the inclusion criteria. They were randomized in a 1:1 ratio to receive teprotumumab or placebo during the 24-week treatment phase and were followed for 1-year of observation. b contains analysis to first response. c illustrates the time course of patients who met response criteria. d demonstrates graded responses at week 24. High response indicates ≥3 mm proptosis reduction and clinical activity score (CAS, 7-point scale) reduced ≥3 points. (From N Engl J Med, Smith TJ, Kahaly GJ, Ezra DG, Fleming JC, Dailey RA, et al. Teprotumumab for thyroid-associated ophthalmopathy, 376; 1748–1761. Copyright © (2017) Massachusetts Medical Society. Reprinted with permission)
Conclusions
Initial results from a multicenter clinical trial suggest that the inhibition of IGF-IR with a specific monoclonal antibody, teprotumumab, might result in substantial improvement of TAO with a favorable safety profile. This therapeutic approach was predicated entirely on observations made at the laboratory bench and thus provided physiological plausibility for the remarkable trial outcomes. A follow-up trial is now being conducted to confirm the encouraging results of the first study. Should those results prove similar to the initial study, it is possible that teprotumumab might replace surgical rehabilitation of the orbital tissues, potentially reducing morbidity and improving the outcomes associated with the disease. Questions concerning whether teprotumumab will be effective in stable TAO must await further investigation.
References
LeRoith D, Werner H, Beitner-Johnson D, Roberts CT Jr. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr Rev. 1995;16:143–63.
Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. 1995;16:3–34.
Hwa V, Oh Y, Rosenfeld RG. The insulin-like growth factor-binding protein (IGFBP) superfamily. Endocr Rev. 1999;20:761–87.
LeRoith DAD, Werner H, Roberts C Jr. Molecular and cellular biology of the insulin-like growth factors. In: Weintraub BD, editor. Molecular endocrinology, basic concepts and clinical correlations. New York, NY: Raven Press; 1995. p. 181–93.
Smith TJ. Insulin-like growth factor-I regulation of immune function: a potential therapeutic target in autoimmune diseases? Pharmacol Rev. 2010;62:199–236.
De Meyts PSW, Palsgaard J, Theede A, Gaugain L, Aladdin H, Whittaker J. Insulin and IGF-I receptor structure and binding mechanism. In: Pessin JE, Saltiel AR, editors. Mechanisms of insulin action. Austin, TX: Landes Bioscience; 2007. p. 1–32.
Pandini G, Frasca F, Mineo R, Sciacca L, Vigneri R, Belfiore A. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J Biol Chem. 2002;277:39684–95.
Slaaby R, Schaffer L, Lautrup-Larsen I, Andersen AS, Shaw AC, Mathiasen IS, et al. Hybrid receptors formed by insulin receptor (IR) and insulin-like growth factor I receptor (IGF-IR) have low insulin and high IGF-1 affinity irrespective of the IR splice variant. J Biol Chem. 2006;281:25869–74.
Baserga R, Sell C, Porcu P, Rubini M. The role of the IGF-I receptor in the growth and transformation of mammalian cells. Cell Prolif. 1994;27:63–71.
Qu X, Wu Z, Dong W, Zhang T, Wang L, Pang Z, et al. Update of IGF-1 receptor inhibitor (ganitumab, dalotuzumab, cixutumumab, teprotumumab and figitumumab) effects on cancer therapy. Oncotarget. 2017;8:29501–18.
Girnita L, Worrall C, Takahashi S, Seregard S, Girnita A. Something old, something new and something borrowed: emerging paradigm of insulin-like growth factor type 1 receptor (IGF-1R) signaling regulation. Cell Mol Life Sci. 2014;71:2403–27.
Smith TJ, Hegedus L. Graves’ disease. N Engl J Med. 2016;375:1552–65.
Adams DD, Purves HD, Sirett NE. The response of hypophysectomized mice to injections of human serum containing long-acting thyroid stimulator. Endocrinology. 1961;68:154–5.
Wang Y, Smith TJ. Current concepts in the molecular pathogenesis of thyroid-associated ophthalmopathy. Invest Ophthalmol Vis Sci. 2014;55:1735–48.
Masetti G, Moshkelgosha S, Kohling HL, Covelli D, Banga JP, Berchner-Pfannschmidt U, et al. Gut microbiota in experimental murine model of Graves’ orbitopathy established in different environments may modulate clinical presentation of disease. Microbiome. 2018;6:97.
Weightman DR, Perros P, Sherif IH, Kendall-Taylor P. Autoantibodies to IGF-1 binding sites in thyroid associated ophthalmopathy. Autoimmunity. 1993;16:251–7.
Rotella CM, Zonefrati R, Toccafondi R, Valente WA, Kohn LD. Ability of monoclonal antibodies to the thyrotropin receptor to increase collagen synthesis in human fibroblasts: an assay which appears to measure exophthalmogenic immunoglobulins in Graves’ sera. J Clin Endocrinol Metab. 1986;62:357–67.
Perros P, Kendall-Taylor P. Biological activity of autoantibodies from patients with thyroid-associated ophthalmopathy: in vitro effects on porcine extraocular myoblasts. Q J Med. 1992;84:691–706.
Tramontano D, Cushing GW, Moses AC, Ingbar SH. Insulin-like growth factor-I stimulates the growth of rat thyroid cells in culture and synergizes the stimulation of DNA synthesis induced by TSH and Graves’-IgG. Endocrinology. 1986;119:940–2.
Pritchard J, Han R, Horst N, Cruikshank WW, Smith TJ. Immunoglobulin activation of T cell chemoattractant expression in fibroblasts from patients with Graves’ disease is mediated through the insulin-like growth factor I receptor pathway. J Immunol. 2003;170:6348–54.
Pritchard J, Horst N, Cruikshank W, Smith TJ. Igs from patients with Graves’ disease induce the expression of T cell chemoattractants in their fibroblasts. J Immunol. 2002;168:942–50.
Douglas RS, Gianoukakis AG, Kamat S, Smith TJ. Aberrant expression of the insulin-like growth factor-1 receptor by T cells from patients with Graves’ disease may carry functional consequences for disease pathogenesis. J Immunol. 2007;178:3281–7.
Douglas RS, Naik V, Hwang CJ, Afifiyan NF, Gianoukakis AG, Sand D, et al. B cells from patients with Graves’ disease aberrantly express the IGF-1 receptor: implications for disease pathogenesis. J Immunol. 2008;181:5768–74.
Cruikshank WW, Berman JS, Theodore AC, Bernardo J, Center DM. Lymphokine activation of T4+ T lymphocytes and monocytes. J Immunol. 1987;138:3817–23.
Schall TJ, Jongstra J, Dyer BJ, Jorgensen J, Clayberger C, Davis MM, et al. A human T cell-specific molecule is a member of a new gene family. J Immunol. 1988;141:1018–25.
Minich WB, Dehina N, Welsink T, Schwiebert C, Morgenthaler NG, Kohrle J, et al. Autoantibodies to the IGF1 receptor in Graves’ orbitopathy. J Clin Endocrinol Metab. 2013;98:752–60.
Krieger CC, Neumann S, Place RF, Marcus-Samuels B, Gershengorn MC. Bidirectional TSH and IGF-1 receptor cross talk mediates stimulation of hyaluronan secretion by Graves’ disease immunoglobins. J Clin Endocrinol Metab. 2015;100:1071–7.
Varewijck AJ, Boelen A, Lamberts SW, Fliers E, Hofland LJ, Wiersinga WM, et al. Circulating IgGs may modulate IGF-I receptor stimulating activity in a subset of patients with Graves’ ophthalmopathy. J Clin Endocrinol Metab. 2013;98:769–76.
Marino M, Rotondo Dottore G, Ionni I, Lanzolla G, Sabini E, Ricci D, et al. Serum antibodies against the insulin-like growth factor-1 receptor (IGF-1R) in Graves’ disease and Graves’ orbitopathy. J Endocrinol Invest. 2018.
Moshkelgosha S, So PW, Deasy N, Diaz-Cano S, Banga JP. Cutting edge: retrobulbar inflammation, adipogenesis, and acute orbital congestion in a preclinical female mouse model of Graves’ orbitopathy induced by thyrotropin receptor plasmid-in vivo electroporation. Endocrinology. 2013;154:3008–15.
Pritchard J, Tsui S, Horst N, Cruikshank WW, Smith TJ. Synovial fibroblasts from patients with rheumatoid arthritis, like fibroblasts from Graves’ disease, express high levels of IL-16 when treated with Igs against insulin-like growth factor-1 receptor. J Immunol. 2004;173:3564–9.
Tsui S, Naik V, Hoa N, Hwang CJ, Afifiyan NF, Sinha Hikim A, et al. Evidence for an association between thyroid-stimulating hormone and insulin-like growth factor 1 receptors: a tale of two antigens implicated in Graves’ disease. J Immunol. 2008;181:4397–405.
Smith TJ, Kahaly GJ, Ezra DG, Fleming JC, Dailey RA, Tang RA, et al. Teprotumumab for thyroid-associated ophthalmopathy. N Engl J Med. 2017;376:1748–61.
Acknowledgements
These studies were supported in part by grant EY08976 from the National Institutes of Health and Core Center for Research grant EY007003 from the National Eye Institute. The author is grateful to Leslie Bordine for her expert assistance in the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author has been issued patents covering his invention of using IGF-I receptor inhibition as a therapy for Graves’ disease, TAO, and other autoimmune diseases. These are held by the Los Angeles Biomedical Institute and the UCLA School of Medicine.
Rights and permissions
About this article

Cite this article
Smith, T.J. The insulin-like growth factor-I receptor and its role in thyroid-associated ophthalmopathy. Eye 33, 200–205 (2019). https://doi.org/10.1038/s41433-018-0265-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41433-018-0265-2
This article is cited by
-
Teprotumumab bei schilddrüsenassoziierter Ophthalmopathie
Der Internist (2020)
