
Abstract
Understanding the role of adaptation in species’ responses to climate change is important for evaluating the evolutionary potential of populations and informing conservation efforts. Population genomics provides a useful approach for identifying putative signatures of selection and the underlying environmental factors or biological processes that may be involved. Here, we employed a population genomic approach within a space-for-time study design to investigate the genetic basis of local adaptation and reconstruct patterns of movement across rapidly changing environments in a thermally sensitive mammal, the American pika (Ochotona princeps). Using genotypic data at 49,074 single-nucleotide polymorphisms (SNPs), we analyzed patterns of genome-wide diversity, structure, and migration along three independent elevational transects located at the northern extent (Tweedsmuir South Provincial Park, British Columbia, Canada) and core (North Cascades National Park, Washington, USA) of the Cascades lineage. We identified 899 robust outlier SNPs within- and among-transects. Of those annotated to genes with known function, many were linked with cellular processes related to climate stress including ATP-binding, ATP citrate synthase activity, ATPase activity, hormone activity, metal ion-binding, and protein-binding. Moreover, we detected evidence for contrasting patterns of directional migration along transects across geographic regions that suggest an increased propensity for American pikas to disperse among lower elevation populations at higher latitudes where environments are generally cooler. Ultimately, our data indicate that fine-scale demographic patterns and adaptive processes may vary among populations of American pikas, providing an important context for evaluating biotic responses to climate change in this species and other alpine-adapted mammals.
Similar content being viewed by others

Introduction
Global biodiversity is declining at an unprecedented rate due to significant anthropogenic impacts on the environment (Ceballos et al. 2015). Since 1900, there have been more than 400 documented vertebrate extinctions (Ceballos et al. 2015), with an additional 515 species currently on the brink (Ceballos et al. 2020). While there are many significant threats, climate change has made one of the most substantial contributions to biodiversity loss (IPCC 2014). Since 1980, global temperatures are estimated to have risen at least 0.7 °C and are projected to rise by at least 1.5 °C by the end of 2100 (IPCC 2014). Climate change affects multiple aspects of biodiversity including species distributions, community composition, and ecosystem functioning, as well as species phenology, genetic composition, and physiology (Bellard et al. 2012). As a result of changing environments, organisms must either move, adjust, adapt, or otherwise face extinction (Lambers 2015; Waldvogel et al. 2020).
Many species extinctions have been attributed to global climate change, which has affected multiple taxa across various geographic regions (Parmesan 2006; Wiens 2016). Alpine species are predicted to be particularly vulnerable to climate fluctuations, as montane ecosystems are sensitive to minor changes in temperature and precipitation, and are geographically isolated (Diaz et al. 2003; La Sorte and Jetz 2010). For montane mammals, both empirical surveys and future projections have revealed evidence of range shifts and losses due to climate change. In general, mammal species range limits have shifted upward, with species at higher elevations undergoing larger range retractions relative to those species that occur at lower elevations (Moritz et al. 2008; Johnston et al. 2012; Moritz and Agudo 2013). While alpine species appear to be moving to suitable climate conditions through geographic range shifts, the extent to which adaptation plays a role in these species’ responses to climate change is less clear (Moritz and Agudo 2013).
Understanding the contribution of adaptation to species responses to climate change is important for assessing population viability and predicting species vulnerability (Moritz and Agudo 2013). In addition to more resource-intensive and logistically challenging methods (e.g., common-garden or reciprocal transplant experiments), population genomics provides a complementary and often more feasible approach to help identify putative signatures of adaptation for threatened vertebrate species (Franks and Hoffmann 2012; Flanagan et al. 2018). Genome scans and genotype-environment association analyses allow for the detection of “outlier” loci that are either significantly differentiated among populations or exhibit significant associations to specified phenotypes or environmental variables (Savolainen et al. 2013). Such analyses of genome-wide variation can provide generate estimates of evolutionary potential and identify loci that may facilitate adaptation to future climate changes (Harrisson et al. 2014). Ultimately, this information can be used to help determine populations or species that require conservation priority, as well as inform adaptive management frameworks for conservation (Flanagan et al. 2018; Hoban 2018). Multiple approaches have been proposed that utilize genomic data to define units for conservation (Funk et al. 2012; Barbosa et al. 2018) and evaluate their vulnerability to climate change (Razgour et al. 2018). Other frameworks have incorporated genomic data into systematic conservation planning to determine priority areas for conservation (Paz-Vinas et al. 2018), as well as to examine the degree to which an existing protected area represents neutral and adaptive evolutionary processes (Hanson et al. 2020).
The American pika (Ochotona princeps) is a montane mammal species considered to be disproportionately affected by climate change (Beever et al. 2003, 2011; Wilkening et al. 2011; Calkins et al. 2012; McCain 2019; Smith 2020). This lagomorph is distributed among patches across western North America where it is restricted primarily to alpine talus slopes found at elevations up to 3000 m above sea level (Smith and Weston 1990). Five intraspecific lineages are currently recognized based on geographic distribution and range-wide genetic variation [Cascade Range, Sierra Nevada, Northern Rocky Mountains, Southern Rocky Mountains, Central Utah; Galbreath et al. 2009; Hafner and Smith 2010]. Dispersal is primarily limited to juveniles that are typically philopatric and remain close to their natal home ranges (Smith and Ivins 1983), though instances of long distance movement (2 km) have been detected using genetic markers (Peacock 1997). American pikas are also thermally sensitive, with elevated basal metabolic rates and high body temperatures relative to other lagomorphs (MacArthur and Wang 1974). As a result, this species is relatively stenothermic and must behaviorally thermoregulate through changes in activity and use of heat avoidance behaviors (Smith 1974b). Altogether, the above characteristics have rendered the American pika an important mammalian species for investigating adaptive capacity and responses to the threats posed by climate change (Beever et al. 2003; Wilkening and Ray 2016).
Many population declines and range retractions in American pikas have been attributed to both historical and contemporary climate changes. On a broad scale, historical climate has influenced the current distribution, with lineage boundaries reflecting glacial climate oscillations since the Last Glacial Maximum (LGM, ~22 kya; Galbreath et al. 2009). At a regional level, several studies report recent population declines within some of the major lineages of the American pika; these apparent extirpations have been linked to climate factors such as temperature (heat and cold stress), precipitation (snowpack, moisture), and elevation (Beever et al. 2003, 2010, 2011, 2016; Erb et al. 2011; Wilkening et al. 2011; Varner and Dearing 2014; Stewart et al. 2015, 2017). Though the variables that constrain the distribution of American pikas differ across geographical areas, climate is hypothesized to have both direct and indirect effects on this species. For instance, climate change may alter the water content or availability of forage, directly increase energetic demands on American pikas, or even alter foraging and dispersal activity, all of which may impact overall persistence (Beever et al. 2003; Erb et al. 2011; Wilkening et al. 2011). In addition to climate sensitivity, low levels of neutral genetic variation have been observed across the species range both at fine-scales within the Cascade lineage (Henry et al. 2012; Russello et al. 2015; Robson et al. 2016) and across multiple other lineages (Klingler et al. 2021), thus raising concerns about the ability for American pikas to adapt to rapid environmental change.
Previous genetic analyses have suggested the presence of local adaptation in American pikas associated with climatic variation (Henry and Russello 2013; Waterhouse et al. 2018). These studies conducted genetic sampling across elevational gradients and employed a space-for-time design, which provides an effective approach for inferring temporal change based upon current patterns of spatial climatic variation, especially when longitudinal studies are not viable (Pickett 1989; Wogan and Wang 2018). Space-for-time designs have been used to evaluate the predictive ability of bioclimatic models (Araújo et al. 2005), as well as model responses to climate change (Blois et al. 2013). In the American pika, Henry and Russello (2013) detected significant associations between allelic variation among amplified fragment length polymorphisms (AFLP) and environmental factors, such as precipitation, snowfall, and maximum temperature across three elevational gradients in Tweedsmuir South Provincial Park (TPP) in British Columbia (BC), Canada. Likewise, Waterhouse et al. (2018) used restriction site-associated DNA sequencing (RADseq; Baird et al. 2008) paired with genotype-environment association analyses across American pika sampling locations spanning two elevational gradients in North Cascades National Park (NOCA), Washington, USA. This study revealed putative signatures of selection at single-nucleotide polymorphisms (SNPs) linked to genes involved in metabolic processes such as oxygen transport, immune function, cell structure, and other processes involved in cold acclimatization. On a broader geographic scale, Rankin et al. (2017) analyzed DNA sequence variation at 50 candidate genes for exemplar individuals spanning the five distinct phylogenetic lineages distributed across western North America, revealing signatures of strong positive selection in genes related to hypoxia responses within liver cells and energy metabolism pathways. Though these studies have provided some evidence of a genetic basis for local adaptation to changing environments in American pikas, they have been limited by the markers used (e.g., AFLPs, Henry and Russello 2013; candidate genes, Rankin et al. (2017), or to specific localities (e.g., NOCA; Waterhouse et al. 2018), impeding our understanding of the broader role and scale at which selective pressures may be operating to promote adaptive responses to climate stress.
Here, we employed a space-for-time design to investigate the extent and distribution of neutral and putatively adaptive SNP genetic variation in American pikas at multiple sites within a distinct phylogenetic lineage. We paired novel RADseq data from the northern extent of the Cascades lineage (TPP) with those previously published from sites at the lineage core in NOCA to examine evidence for local adaptation across elevational gradients located ~550 km apart. We further annotated robust outlier loci to candidate genes exhibiting putative signatures of selection within and among transects and explored their biological implications. Lastly, we compared patterns of genetic diversity, structure, and migration within and among transects to infer fine-scale demographic patterns and contrast how these patterns vary spatially across the Cascade lineage of American pikas. We hypothesized that American pikas would exhibit: (1) limited dispersal within and among transects as reflected in patterns of neutral genetic variation; and (2) significant genotype-environment associations within and among transects linked to genomic regions related to climate stress.
Materials and methods
Sample collection
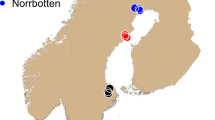
A total of 49 individuals were sampled along a previously established elevational transect ranging from 230 to 1480 m above sea level within TPP (Fig. 1B). Following Waterhouse et al. (2018), individuals were live-trapped, and ear tissue was collected at six sampling sites (A1, A3, B–E) along the Hill transect within TPP, as well as opportunistically at two supplemental sites (A4, TT; Fig. 1B). All trapping and tissue sampling were conducted in accordance with University of British Columbia Animal Care Protocol # A15-0280 and BC Ministry of Environment and Climate Change Strategy Permit # 102801.

A Range-wide distribution (redrawn from Galbreath et al. 2009). Bold lines indicate genetic lineages. Diamonds indicate regions sampled. B Topographic map of sampling distribution of the Hill transect in TPP. C Topographic map of sampling distribution along TL and PP transects in NOCA.
Climate data
Data for 28 directly calculated climate variables were downloaded from ClimateWNA (Wang et al. 2012) for 1981–2010 at each sampling site based upon latitude, longitude, and elevation (Supplementary Table S1). To reduce collinearity among the climate variables, we ran a PCA on centered and scaled climate data for each transect using the prcomp function in R studio (R Core Team 2019). PC axes that explained at least 98% of the variation in climate among the sampling sites within each transect were then used as composite climate variables.
Genomic data collection
We used RADseq to collect genotypic data for the 49 individuals from the Hill transect in TPP following Baird et al. (2008) as modified in Lemay and Russello (2015). Briefly, 500 ng of each DNA sample was digested with SbfI, and P1 identifying barcodes were ligated to each sample. Next, barcoded samples were pooled, sheared using a Biorupter® (Diagenode, Liège, Belgium), and size selected for 300–600 bp fragments using a Pippin Prep (Blue Sage Science, Beverly, MA, USA). After size selection, the retained DNA fragments underwent P2 adapter ligation followed by PCR amplification. The resulting library was size-selected again for 300–650 bp fragments and sequenced using a single lane of Illumina HiSeq 2500 paired-end 100 bp sequencing at the McGill University and Génome Québec Innovation Centre. In addition, previously published (Waterhouse et al. 2018) raw sequence reads generated via SbfI RADseq for 59 individuals sampled at eight main sites (PP01-04; TL01-04) and five supplementary sites (PPTrl, PPHwy, PP1.5,TL0.5, TL0.7) along the Pyramid Peak (PP) and Thornton Lakes (TL) transects within NOCA (Fig. 1C) were downloaded from the NCBI SRA (#SRP136538).
Reference alignment and SNP genotyping
Using the process_radtags module of the Stacks 2.0 pipeline (Rochette et al. 2019), sequencing reads from the Hill transect were demultiplexed, trimmed to 94 bp, and reads with uncalled bases were removed (-c). We combined these new data with the sequence reads previously collected across the PP and TL transects in NOCA (108 individuals total) and aligned them to the O. princeps reference genome (OchPri4.0, GenBank Assembly ID: GCA_014633375.1; Sjodin et al. 2021) using BWA-mem (Li 2013) with default parameters. The resulting bam files were sorted and then processed with the gstacks module of the Stacks 2.0 pipeline to simultaneously identify and genotype SNPs. Next, the populations module was run to filter and generate a SNP dataset after performing a parameter sensitivity analysis (Supplementary Table S2) and removing any individuals that had a mean sequencing depth <6×, as well as those from sites with <3 samples. From the sensitivity analysis, we chose a middling set of parameters that aimed to balance the amount of missing data and sequencing depth across all included individuals and would allow us to retain one SNP per RADtag (–write-single-snp), present in 80% of all individuals (-R 0.8), with a minor allele frequency of at least 0.03 (–min-maf 0.03; Supplementary Table S2). The subsequent dataset was then filtered with VCFtools (Danecek et al. 2011) to retain biallelic SNPs with a minimum depth of 10× and a maximum depth of 100× following Waterhouse et al. (2018).
Prior to outlier SNP detection and population genomic analysis, the initial filtered dataset was split into transect-specific datasets. For each of these datasets, we removed SNPs that were monomorphic, invariant, or had more than 20% missing data. We also estimated pairwise relatedness for each dataset using the program COANCESTRY as implemented in the R package “related” (Pew et al. 2015), removing one individual from each pair considered first-order relatives (Rxy ≥ 0.5). This enabled us to limit potential bias resulting from the presence of highly related individuals within the data (Astle and Balding 2009).
Outlier detection and annotation
We employed a combination of three different outlier detection methods to help limit the rate of false positive detections, which can result from genome scans and genotype-environment association analyses (Rellstab et al. 2015). First, we used Bayescan v 2.0 (Foll and Gaggiotti 2008), which calculates the probability that a SNP locus is under selection based upon levels of genetic differentiation (Fst) relative to those expected under neutral conditions. This analysis was run using 100,000 iterations, with a burn-in of 50,000 iterations, and prior odds value of 10. We conducted five independent runs for each transect initialized with a different random seed every run. SNP loci with median q values <0.2 across all five runs for a given transect were considered outliers.
Next, we identified outlier loci using latent factor mixed modeling (LFMM; Frichot et al. 2013) as implemented in the R package LEA v 2.8.0 (Frichot and François 2015). This method tests for significant associations between allele frequencies at each SNP locus and selected environmental variables with one or more latent factors accounting for underlying population structure. To determine the optimal number of latent factors (K), sparse non-negative matrix factorization (snmf) analyses were conducted for each transect as implemented in LEA. We ran snmf for ten repetitions from K = 1–10 and then selected the K value with the minimum cross entropy. Next, we imputed missing data based upon the chosen K value using the impute function in the LEA package to increase statistical power of the association analysis (Browning 2008) as recommended by the authors. Then, we ran the lfmm function on each imputed dataset with 10 repetitions, 100,000 iterations, and a burn-in of 50,000 iterations for all composite climate variables. The resulting z scores for all loci were combined across all ten runs, and p values were adjusted after calibrating the genomic inflation factor manually as described in the LEA package manual. SNPs with significant associations to each climate variable were identified as outliers after Benjamini–Hochberg correction for a false discovery rate (q) of 0.05.
Finally, we identified significant correlations between environmental variables and population allele frequencies at each SNP while accounting for underlying associations among populations using BayPass v 2.1 (Gautier 2015). First, we ran the core model to create a covariance matrix among population allele frequencies within each transect. This model was run using 20 pilot runs of 10,000 iterations, with a burn-in of 50,000 iterations, sampling every 25 iterations. Using these same parameters, we then conducted five runs of the auxiliary model using the output of the core model to help account for underlying population structure and test for associations with environmental variables. We then calculated the median Bayes Factor (BF) for each locus across all five runs of the auxiliary model. Those SNPs with a BF of 20 decibans (dB) and higher were considered outliers.
SNPs identified as outliers either by two or more detection methods within each transect, or in multiple transects using any detection method were combined into a single dataset (hereafter referred to as the “robust outlier dataset”). All pairs of loci were then tested for evidence of linkage disequilibrium (LD), using an exact test with 10,000 dememorization steps, 100 batches, and 10,000 iterations per batch as implemented in GENEPOP v 4.7.0 (Rousset 2008). We removed one locus from each pair exhibiting LD (p < 0.05) after performing a Benjamini–Hochberg correction using the p.adjust function in R (R Core Team 2019). To determine the putative biological function of the robust outliers, we conducted NCBI blastn (Zhang et al. 2000) sequence similarity searches of the nucleotide (nt) database using the 200 base pair reference genome sequence centered around each high-confidence outlier SNP, employing a word size of 11 and an e-value cut-off of 1e–10. Results were mapped and annotated using BLAST2GO (Götz et al. 2008).
Population genomic analyses
To generate a neutral SNP dataset for the population genomic analyses, any locus that was monomorphic within transects or identified as an outlier by any method in any transect was removed from the initial filtered dataset generated across all sampled individuals. This dataset was further filtered by testing for Hardy–Weinberg Equilibrium (HWE) within each sampling site using the -hardy function in VCFtools v 0.1.17 (Danecek et al. 2011). SNPs that significantly deviated from HWE (p < 0.05) in at least 50% of the main sampling sites among all three transects were also excluded. The retained dataset was then used to conduct population genomic analyses.
We estimated measures of genetic diversity including the effective number of alleles (Ae), observed and expected heterozygosity (Ho, He), and inbreeding coefficients (Gis) for each sampling site using GenoDive v 3.03 (Meirmans and Van Tienderen 2004). Pairwise relatedness within each sampling site was calculated using the program COANCESTRY as implemented in the R package “related” (Pew et al. 2015). We employed the Ritland (1996) estimator as it has been shown to be the least sensitive to allelic dropout and genotyping error (Attard et al. 2018).
To examine patterns of genetic differentiation, values of pairwise θ (Weir and Cockerham 1984) were calculated among sampling sites for both the neutral dataset and the transect-specific outlier datasets using GenoDive (Meirmans and Van Tienderen 2004). Significance was assessed across 10,000 permutations. Individual ancestry was estimated using a Bayesian clustering analysis as implemented in ADMIXTURE v 1.3.0 (Alexander et al. 2009). We ran the program from K = 1–10 with 10 iterations at each value of K with cross-validation (CV) enabled. The value of K with the lowest mean (CV) error across all ten runs was considered optimal following author recommendations (Alexander et al. 2015; Supplementary Fig. S1). Because strong genetic structure across populations can mask within-population structure, we then ran ADMIXTURE for each transect separately from K = 1–10 using the above parameters. In addition, each of the transect-specific outlier datasets was run through ADMIXTURE to further examine spatial patterns of genetic structure along each elevational gradient. Based upon previous studies, we considered there to be no admixture when individuals had proportions of ancestry to other populations that were less than 25% (Latch et al. 2006, 2008; Holbrook et al. 2012).
Detection of directional migration
We used the BA3-SNPs v 3.0.4 reconfiguration of BayesAss3 (Mussmann et al. 2019) to determine the amount and direction of migration within each transect. For each transect-specific neutral dataset, BA3-SNPs was run five times using a different random seed across 10,000,000 iterations with a burn-in of 1,000,000 iterations and sampling every 100 iterations. Optimal model mixing parameters (-a,-m, -f) were determined for each dataset using the BA3-SNPS-autotune program (Mussmann et al. 2019). We then assessed model convergence across all five runs and calculated median migration estimates with 95% credible sets (median ± 1.96*SD) as suggested by the original authors (Wilson and Rannala 2003). Those estimates where the accompanying credible set did not include zero were considered to represent significant migration. We further examined patterns of migration along each transect by performing assignment tests on the four individuals sampled at supplementary sites relative to the main sites using the estimator of Rannala and Mountain (1997) as implemented in GeneClass2 v 2.0 (Piry et al. 2004). For this analysis, we used a random subset of 5000 SNPs from each of the transect-specific neutral datasets due to computational limitations of the software package.
Results
Climate data
For all three transects, the first two PC axes were retained as composite climate variables. In the Hill transect, PC1 and PC2 explained 96.88 and 2.53% of the variation in climate along the elevational gradient, while PC1 explained 90.79 and 95.79% of the variation in PP and TL, and PC2 explained an additional 7.98 and 4.13%, respectively. Generally, PC1 was significantly correlated with elevation in all transects; however, this relationship was positive in PP (r = 0.998, n = 4, p = 1.98E–2) and TL (r = 0.998, n = 4, p = 1.78E–2), and negative in Hill (r = –0.997, n = 6, p = 1.30E–05). In the Hill transect, PC1 was associated with temperature and solar radiation, while PC2 was associated with precipitation. In both NOCA transects (PP and TL), PC1 was representative of precipitation and temperature, while PC2 mainly reflected solar radiation (Supplementary Table S3).
Reference alignment and SNP genotyping
Overall, 437,166,300 sequencing reads were generated across the combined datasets with a mean of 4,047,836 (SD = 1,234,069) reads per individual. Across all individuals, a mean of 97.96% (SD = 0.42) of reads aligned to the reference genome. After initial filtering, 49,074 SNPs were genotyped across 97 individuals from the 14 main sampling sites with a mean of 2.39% (SD = 1.58) missing data and a mean depth of 24.34× (SD = 6.67) per individual. Within each transect there were 22,786, 8706, and 9572 SNPs that were either invariant, monomorphic, or missing in Hill, PP, and TL, respectively, and were flagged for removal (Supplementary Table S4). This resulted in datasets of 26,288 SNPs for outlier detection in the Hill transect, 40,368 SNPs in the PP transect, and 39,502 SNPs in the TL transect.
Outlier detection and annotation
Within the Hill transect, a total of 2328 outlier SNPs were identified across all three outlier detection methods (Table 1). For the LFMM analysis, snmf identified an optimal K = 3 (Supplementary Fig. S2). Bayescan detected 54 outliers, while the LFMM and BayPass analyses detected 1709 and 565 outliers, respectively (Table 1 and Fig. 2A). In total, 1131 of these loci were significantly associated with PC1, while 1143 were significantly associated with PC2; 91 were associated with both composite climate variables (Table 1). Overall, there were 240 high-confidence outliers found across multiple detection methods, 18 of which were identified with all three analyses (Fig. 2A).

Results of outlier detection within the Hill (A), Pyramid Peak (PP; B), Thornton lakes (TL; C), and comparison of outliers detected among all three transects with any detection method (D). Total counts of outlier loci detected with each method for A–C or within each transect (D) are included in parentheses. Intersecting regions between analytical approaches or transects indicated robust outliers.
In the PP transect, a total of 2008 outlier SNPs were identified across all outlier detection methods (Table 1). For the LFMM analysis, snmf identified an optimal K = 1 (Supplementary Fig. S2). Bayescan detected 60 outliers, while the LFMM and BayPass analyses detected 230 and 1718 outliers, respectively (Table 1 and Fig. 2B). In total, 827 outliers were significantly associated with PC1 and 1121 were associated with PC2, with five of these outliers associated with both composite climate variables (Table 1). Overall, there were 204 high-confidence outliers detected in the PP transect, with 30 of those outliers identified using all three detection methods (Fig. 2B).
Along the TL transect, there were 2679 outlier SNPs detected across all methods. For the LFMM, snmf identified an optimal K = 1 (Supplementary Fig. S2). Bayescan detected 50 outliers, while the LFMM and BayPass analyses detected 383 and 2246 outliers, respectively (Table 1 and Fig. 2C). In total, 1190 outliers were significantly associated with the PC1 environmental variable, 1439 were associated with PC2, and 3 were associated with both composite climate variables. Altogether, there were 322 high-confidence outlier loci detected with multiple methods in TL, 37 of which were detected by all three methods (Fig. 2C).
In total, the robust outlier dataset consisted of 964 SNPs. In addition to the high-confidence outliers detected within each transect (Fig. 2), there were 198 SNPs identified as outliers with at least one method in multiple transects, one of which was detected in all three transects (Fig. 2D), and 65 of which were also high-confidence within a transect, leaving 899 unique robust outlier SNPs. Among all robust outliers, no pairs of loci were in LD. Of the 899 robust outlier SNPs, 101 had BLASTn matches, 46 of which were specific to O. princeps and corresponded to genes of known function (Supplementary Table S5). Despite the lack of statistically significant LD, there were four instances where multiple SNPs annotated to the same gene; in three of these cases the SNPs associated with the same gene were found on the same scaffold of the reference genome (Supplementary Table S5). Common gene ontology (GO) functions included, but were not limited to, ATP binding, ATP citrate synthase activity, ATPase activity, hormone activity, metal ion binding, and protein binding (Supplementary Table S5).
Population genomic analyses
After the removal of all outlier, monomorphic, and invariant SNPs detected in at least one transect from the initial filtered dataset (n = 31,815), 17,259 polymorphic SNPs remained for neutral genetic analyses; none of these deviated from HWE in ≥50% of the main sampling sites. Across all 14 sampling sites, Ho estimates ranged from 0.170 in C along the Hill transect to 0.379 in the PP02 site (Table 2). For both NOCA transects (PP and TL), estimates of diversity were consistent with those found in Waterhouse et al. (2018). Most sampling sites had evidence for a slight heterozygote excess and outbreeding except for PP01, TL02, and TL03. These latter sites each exhibited a slight heterozygote deficit, with Gis estimates that were positive and significantly different from zero (Table 2). Overall, values of Gis ranged from –0.213 in A3 to 0.061 in PP01, while within-site relatedness ranged from –0.040 in B to 0.182 in TL04 (Table 2).
Neutral pairwise θ estimates ranged from 0.009 to 0.524 among sites within and among transects (Table 3). Within transects, θ ranged from 0.009 to 0.076 in Hill, 0.029 to 0.070 in PP, and 0.036 to 0.111 in TL (Table 3). All pairwise comparisons between sampling sites were significant (p < 0.05) except for those between A1–A3, B–D, PP01–PP03, PP02–PP03, and PP03–PP04. Outlier pairwise θ estimates among sites ranged from 0.040 to 0.401 within Hill, 0.255 to 0.423 in PP, and 0.158 to 0.718 within TL (Table 3). All pairwise comparisons were significant (p < 0.05).
For the ADMIXTURE analyses, we found an optimal K = 3 across all transects using the neutral dataset (Supplementary Fig. S1D). Each cluster corresponded to a single transect, with only minimal admixture detected between PP and TL (Fig. 3). We found no evidence for within-transect structure using the neutral dataset, with an optimal K = 1 selected for all three transects (Supplementary Fig. S1). ADMIXTURE analyses using the transect-specific outlier datasets found optimal K of 4, 2, and 3 for the Hill, PP, and TL transects, respectively (Supplementary Fig. S1). Within the Hill transect, there was evidence for distinct genetic groups corresponding to A1, A3, C, and E (Fig. 3). B and D were found to exhibit admixture between A1 and C, and C and E, respectively (Fig. 3). Within the PP transect, PP01 and PP04 each represented a unique genetic cluster, while PP02 and PP03 exhibited varying degrees of admixture between the PP01 and PP04 sites (Fig. 3). Within the TL transect, distinct genetic groups were found for TL01, TL02, and TL04, with admixture among the TL02 and TL04 sites in TL03 (Fig. 3).

The number of outlier SNPs within each transect is shown above the “Outlier” plot.
Directional migration
Across all three transects, patterns of migration were primarily downslope in PP and TL, with some evidence for upslope migration within Hill. Migration rates within the Hill transect were significant from C to all other sample sites, while migration rates were significant from PP04 to PP02 along PP and from TL03 to TL02 in TL (Table 4). All other migration rates were not significant. Individual pikas sampled at supplementary sites along each transect were all assigned to main sites with 100% assignment accuracy; within the Hill transect, all four individuals assigned to the neighboring sites, while all individuals from the NOCA transects assigned to upslope populations (Supplementary Table S6).
Discussion
Identifying environmental factors and biological processes that may influence adaptation is important for evaluating evolutionary potential and predicting species’ responses to climate change (Hoffmann and Sgrò 2011). Using a space-for-time design, we investigated genomic diversity, gene flow, and spatial genetic structure in American pikas along three independent elevational transects across which environmental conditions changed rapidly at the northern extent and core of the Cascades lineage. Our results revealed varied patterns of putative local adaptation to environmental variables linked to climate stress along each elevational gradient, as well as evidence for limited directional migration. Collectively, our findings suggest that patterns of migration may be constrained by rapidly changing environmental conditions, which could be especially problematic for alpine species distributed across naturally fragmented landscapes.
Robust outlier detection and functional annotations
Of the 899 robust outlier SNPs detected, ~22% were identified by at least one detection method in multiple transects (n = 198), and one was identified in all three transects, lending confidence to their status as candidate loci under selection. Of these, 46 SNPs were annotated to genes of known function in the American pika, several of which could have relevance to environmental selection. Recovered annotations matched to genes such as ATP-citrate lyase (ACLY), chloride voltage-gated channel 2 (CLCN2), kinesin family member 21B (KIF21B), and leucine rich repeat containing 8 VRAC subunit E (LRRC8E) (Supplementary Table S5). Though the exact functions of these genes in O. princeps are not known, several of them have been associated with various cellular responses to stress in other organisms. For instance, ACLY is an enzyme involved in energy metabolism in mammalian cells that helps to synthesize acetyl-CoA, which is a precursor for fatty acid synthesis (Chypre et al. 2012). Recent research has identified ACLY as part of a response to water stress in the kidney cells of camels; downregulation of this gene was linked to the prevention of cell dehydration as well as the slowing of cellular respiration and metabolism (Zhang et al. 2020). In human embryonic kidney cells, CLCN2 channel activation has been linked to heat shock proteins Hsp70 and Hsp90 (Hinzpeter et al. 2006). Hsp90 is associated with increased activity of CLCN2 that may reflect responses to heat and oxidative stress (Hinzpeter et al. 2006). Likewise, LRRC8E has been identified as one of five proteins in the LRRC8 complex that is involved in volume-regulated anion channel functioning in mammalian cells, which regulates cell volume in response to osmotic changes (Jentsch et al. 2016). These protein subunits have been shown to be directly modified by oxidative stress; LRRC8E is one subunit in particular that is activated by oxidative processes (Gradogna et al. 2017). Overall, the functional annotations detected in this study are consistent with previous results that suggest selection may be acting upon genes related to heat stress, oxygen transport, and other metabolic processes in American pikas (Rankin et al. 2017; Waterhouse et al. 2018).
It is important to note that the high-confidence outlier loci identified across transects in this study may also be associated with the post-glacial colonization history of the American pika, as all three of these transects are part of the Cascades lineage (Fig. 1). During the LGM, the extent of the Cordilleran ice sheet in North America reached as far south as the 49th parallel, effectively covering the geographic area surrounding TPP (Galbreath et al. 2009). As climate conditions started to warm, American pikas expanded northward from refugia in the Southern US (Galbreath et al. 2009). Given this evidence for recent demographic expansion into southern BC following glacial retreat, it is possible that American pikas along the Hill transect in TPP share ancestral polymorphisms with populations in TL and PP. Therefore, outliers that are common to multiple transects in this study may not reflect independent responses to selection, but rather shared alleles due to historical climate adaptation throughout the entire Cascades lineage. Because shared ancestry, parallel mutations, and hybridization can all lead to signatures of convergence among populations, challenges arise when disentangling the relative influence of each process on observed genomic variation (Stern 2013; Lee and Coop 2017, 2019). Future research could employ coalescent models to investigate mechanisms of adaptation to climate in American pikas and perhaps provide insights on the likelihood of similar evolutionary adaptations occurring independently elsewhere across the range.
Patterns of genetic diversity and demography
We found consistently low levels of genetic diversity across all sites that may reflect inbreeding, recent population declines, or in the case of the Hill transect, range expansion. In PP01, TL02, and TL03, we detected evidence for a heterozygote deficit that may be the result of inbreeding (Table 2). The other sampled populations exhibited a slight heterozygote excess, but showed no evidence of inbreeding or high within-population relatedness, except for TL04, which had the highest level of within-site relatedness (Table 2). These instances of heterozygote excess may indicate population decline due to recent bottleneck events (Cornuet and Luikart 1996). Previous genetic work conducted on American pikas along the Hill transect revealed evidence for recent population decline (Henry et al. 2012). Using microsatellite genotypic data, Henry et al. (2012) detected excess heterozygosity and shifts in allele frequency distributions at multiple sites at various elevations, both of which are indicative of population contraction. While our results are consistent with population decline along each transect, we did not explicitly investigate signatures of a bottleneck using the conventional heterozygote excess test (Cornuet and Luikart 1996) as it does not employ the infinite-sites mutation model that is most appropriately applied to SNPs (Kimura 1971; Morin et al. 2004). In addition, small per-site sample sizes precluded the use of Bayesian coalescent analyses to infer changes in effective population sizes over time (Heled and Drummond 2008).
Within the Hill transect, estimates of heterozygosity (Table 2) may reflect the post-glacial expansion of American pikas after the LGM (Galbreath et al. 2009). Expansion from unglaciated areas would lead to lower diversity in these more recently colonized populations (Hewitt 19962000; Excoffier et al. 2009). As American pikas had already occupied the geographical area surrounding NOCA by the height of the LGM (Hafner and Sullivan 1995), further spread into TPP following glacial retreat likely resulted in low diversity as a consequence of repeated founder events (Angert et al. 2020). Moreover, populations toward range margins are hypothesized to have lower genetic diversity relative to those toward the range core (Eckert et al. 2008). Our results are consistent with this prediction as American pikas at the northern-most extent of the range in TPP exhibited lower diversity than those at more southerly latitudes in NOCA.
Overall, estimates of inbreeding and relatedness within each transect provide evidence for some mechanism that allows American pikas to avoid inbreeding. Previous studies in the Columbia River Gorge and the Sierra Nevada regions using microsatellite genotypic data detected patterns that indicated mating between American pikas of intermediate relatedness (Peacock and Smith 1997; Robson et al. 2016). In contrast, our study based on genome-wide SNPs found relatedness values that were generally low and little evidence for inbreeding within sites (Table 2). These results aligned with those from collared pikas (Ochotona collaris), the sister species to American pikas, which suggested they are capable of dispersing far enough to avoid mating with relatives due to their lower population densities (Zgurski and Hik 2012). As dispersal in American pikas is largely driven by competition for habitat (Peacock 1997), it could be that population densities along each transect may be lower than in other areas of pika occurrence, and/or these transects may have greater amounts of suitable habitat than in more southerly areas.
Influence of regional climate on genetic structure and directional migration
As climate change proceeds, it is predicted that species ranges will move upslope where habitat may be more suitable for cold-adapted species (Johnston et al. 2012), a phenomenon previously documented in the American pika (Beever et al. 2003, 2011). Here, we found evidence for varied genetic structure and directional movement along three elevational transects sampled at the northern extent and range core of the Cascades lineage of the American pika. For all three transects, there was no evidence for population structure at neutral loci (Supplementary Fig. S1) and estimates of divergence were low (θ; Table 3), indicating some degree of gene flow among sampling sites. Notably, the distribution of outlier variation along each transect was considerably more structured than at neutral loci (Fig. 3), with higher estimates of θ (Table 3). Altogether, these patterns of differentiation and population clustering suggest that genomic signatures of putative adaptation differ between populations at high and low elevations, and that the transfer of adaptive variation may be limited.
Within NOCA, there was significant downslope migration detected in both transects [PP04 (1580 m) to PP02 (820 m), TL03 (1390 m), and TL02 (780 m); Table 4]. Results of individual assignment tests also revealed evidence for downslope migration (Supplementary Table S6). All three individuals sampled at supplementary sites within PP (330–615 m) assigned to the highest elevation site PP04 (1580 m), while the individuals sampled at TL05 (150 m) and TL07 (265 m) in TL assigned to TL03 (1390 m) and TL02 (780 m), respectively. These patterns are similar to those previously reported in Waterhouse et al. (2018) and may reflect demographic influences on dispersal in NOCA; field observations indicated that high elevation sites were more densely populated (M. Waterhouse, personal observation). Adult pikas are highly territorial and often defend large territories within a talus patch (Kilham 1958). As juveniles mature, they may be forced to disperse if a vacant territory is not available in a density-dependent process (Smith 1974a). It is possible that higher elevation sites in NOCA will continue to be densely populated even with climate change, while lower elevation sites become increasingly unsuitable. This could potentially create a source-sink dynamic where high elevation source sites contribute migrants maladapted to environmental conditions at low elevation sites. In contrast, results from the Hill transect revealed generally upslope migration from source sites at lower elevations. Significant migration estimates were identified from C (362 m) to all other sites along the Hill transect, and the magnitude of these values was higher than in the PP and TL transects (Table 4). In addition, assignment tests classified individuals from the supplementary TT site (230 m) as migrants from C (362 m) and D (301 m) (Supplementary Table S6). As such, the differences in migration patterns detected among the broader areas in this study may reflect an increased propensity for American pikas to disperse among lower elevation populations at higher latitudes, which is counter to what has been found at more southerly locations (Castillo et al. 20142016).
Regional differences in temperature and precipitation may help explain these contrasting patterns of American pika movement across the Cascades lineage. For example, the environment in TPP is generally cooler compared to the more southerly NOCA sites (Supplementary Table S1). American pikas are highly dependent upon talus for behavioral thermal regulation (Moyer‐Horner et al. 2015). However, this dependency may be relaxed in a cooler environment, which would facilitate dispersal through intervening habitat between talus patches (Chapman and Flux 1990). Our results are further consistent with previous studies that suggest that American pika distribution is affected by water balance and precipitation (Beever et al. 2010, 2011; Erb et al. 2011). Across transects, mean annual precipitation (MAP) had a much lower rate of change per meter of elevation along the Hill transect compared to either of the NOCA transects (∆MAPHill = 0.181, ∆MAPPP = 0.772, ∆MAPTL = 0.674; Supplementary Table S1). A similar pattern was also observed for values of winter, spring, and autumn precipitation (Supplementary Table S1). Thus, the relative stability in precipitation values along the Hill transect may also encourage more frequent and successful dispersal events for American pikas in this more northerly geographic area. In contrast, recent work in NOCA detected decreases in American pika abundance at low elevation sites (<1400 m) that were attributed to amounts of snowpack and vapor pressure deficit (VPD; Johnston et al. 2019). While the climate variables used in the current study did not include VPD or snowpack explicitly, measures of winter precipitation along the PP and TL transects were 1.48× and 1.37× greater at the highest elevation sites (Supplementary Table S1). Given that snowpack has been found to provide pikas with insulation against harsh winter temperatures (Morrison and Hik 2007), the relatively large difference in winter precipitation across elevations within NOCA may be impeding movement between sites. Other studies across the range have indicated that American pika occurrence is influenced by snowpack (Beever et al. 2010; Yandow et al. 2015), however, this impact may not be universal, as survivorship was not affected by snowfall in some areas such as the Sierra Nevada (Smith and Millar 2018). Additional studies involving a broader sample distribution across the Cascades lineage, as well as genotype-environment association analyses with more specific temperature and precipitation climate variables would be required to further test these hypotheses and assess patterns of migration across a latitudinal cline.
Taken together, our data suggest that fine-scale demographic patterns and adaptive processes may vary among populations of American pikas as climate changes. We found limited directional migration among populations along elevational gradients, indicating that dispersal may be directed by habitat suitability and availability. Moreover, we found signatures of putative adaptation along each elevational gradient, including shared outlier loci across transects that annotated to genes potentially associated with response to climate stress. The presence of these patterns across multiple geographic regions may indicate that American pikas are experiencing climate-mediated selection. In addition, evidence for primarily downslope migration in NOCA suggests that future range retraction in the region may be due to extirpations of lower elevation populations rather than direct movement upslope in response to warming conditions (Waterhouse et al. 2018). Conversely, American pikas at higher latitudes within TPP may harbor greater mobility as the climate changes; though limited, the migration of individuals upslope in the Hill transect may indicate that adaptations resulting from climate stress in populations at lower elevations will be able to shift upward as climate changes. This dynamic could provide high elevation populations with some resistance to warmer conditions through evolutionary rescue (Carlson et al. 2014). Due to the limited spatial scale of this study, we are unable to generalize our findings beyond the Cascades lineage of American pikas; however, low levels of standing genomic variation across several other genetic lineages (Klingler et al. 2021) warrant further examination of the adaptive capacity of this species. Forthcoming population genomic studies will expand upon existing work to test hypotheses related to putative adaptation and parallel selection across the entire range of the American pika.
Despite some limitations, our analyses provide important context for evaluating wildlife movement and adaptation in response to climate change. Here, we demonstrate that putatively adaptative genomic responses differ across geographical regions and environmental conditions at a fine scale, indicating that climate change may affect populations of species unequally. Our results also suggest that patterns of migration may potentially be constrained by rapidly changing environmental conditions, especially in warmer climates. This could be especially problematic for alpine species distributed across naturally fragmented landscapes, where metapopulation connectivity is critical for the maintenance of genetic diversity (Rubidge et al. 2012).
Data availability
All Illumina raw reads are available from the NCBI sequence read archive (BioProject ID: PRJNA761027); RAD tag sequences and SNP genotypic data are deposited in DRYAD (https://doi.org/10.5061/dryad.w0vt4b8s7).
References
Alexander DH, Novembre J, Lange K (2009) Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19:1655–1664
Alexander DH, Shringarpure SS, Novembre J, Lange K (2015) Admixture 1.3 software manual. UCLA Hum Genet Softw Distrib, Los Angeles
Angert AL, Bontrager MG, Ågren J (2020) What do we really know about adaptation at range edges? Annu Rev Ecol Evol Syst 51:341–361
Araújo MB, Pearson RG, Thuiller W, Erhard M (2005) Validation of species–climate impact models under climate change. Glob Change Biol 11:1504–1513
Astle W, Balding DJ (2009) Population structure and cryptic relatedness in genetic association studies. Stat Sci 24:451–471
Attard CRM, Beheregaray LB, Möller LM (2018) Genotyping-by-sequencing for estimating relatedness in nonmodel organisms: Avoiding the trap of precise bias. Mol Ecol Resour 18:381–390
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA et al. (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One 3:1–7
Barbosa S, Mestre F, White TA, Paupério J, Alves PC, Searle JB (2018) Integrative approaches to guide conservation decisions: Using genomics to define conservation units and functional corridors. Mol Ecol 27:3452–3465
Beever EA, Brussard PF, Berger J (2003) Patterns of apparent extirpation among isolated populations of pikas (Ochotona princeps) in the Great Basin. J Mammal 84:37–54
Beever EA, Ray C, Mote PW, Wilkening JL (2010) Testing alternative models of climate-mediated extirpations. Ecol Appl 20:164–178
Beever EA, Ray C, Wilkening JL, Brussard PF, Mote PW (2011) Contemporary climate change alters the pace and drivers of extinction. Glob Change Biol 17:2054–2070
Beever EA, Perrine JD, Rickman T, Flores M, Clark JP, Waters C et al. (2016) Pika (Ochotona princeps) losses from two isolated regions reflect temperature and water balance, but reflect habitat area in a mainland region. J Mammal 97:1495–1511
Bellard C, Bertelsmeier C, Leadley P, Thuiller W, Courchamp F (2012) Impacts of climate change on the future of biodiversity. Ecol Lett 15:365–377
Blois JL, Williams JW, Fitzpatrick MC, Jackson ST, Ferrier S (2013) Space can substitute for time in predicting climate-change effects on biodiversity. Proc Natl Acad Sci USA 110:9374–9379
Browning SR (2008) Missing data imputation and haplotype phase inference for genome-wide association studies. Hum Genet 124:439–450
Calkins MT, Beever EA, Boykin KG, Frey JK, Andersen MC (2012) Not-so-splendid isolation: modeling climate-mediated range collapse of a montane mammal Ochotona princeps across numerous ecoregions. Ecography 35:780–791
Carlson SM, Cunningham CJ, Westley PAH (2014) Evolutionary rescue in a changing world. Trends Ecol Evol 29:521–530
Castillo JA, Epps CW, Davis AR, Cushman SA (2014) Landscape effects on gene flow for a climate-sensitive montane species, the American pika. Mol Ecol 23:843–856
Castillo JA, Epps CW, Jeffress MR, Ray C, Rodhouse TJ, Schwalm D (2016) Replicated landscape genetic and network analyses reveal wide variation in functional connectivity for American pikas. Ecol Appl 26:1660–1676
Ceballos G, Ehrlich PR, Raven PH (2020) Vertebrates on the brink as indicators of biological annihilation and the sixth mass extinction. Proc Natl Acad Sci USA 117:13596–13602
Ceballos G, Ehrlich PR, Barnosky AD, García A, Pringle RM, Palmer TM (2015) Accelerated modern human–induced species losses: entering the sixth mass extinction. Sci Adv 1:e1400253
Chapman JA, Flux JE (1990) Rabbits, hares and pikas: status survey and conservation action plan. IUCN.
Chypre M, Zaidi N, Smans K (2012) ATP-citrate lyase: a mini-review. Biochem Biophys Res Commun 422:1–4
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA et al. (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158
Diaz HF, Grosjean M, Graumlich L (2003) Climate variability and change in high elevation regions: past, present and future. Clim Change 59:1–4
Eckert CG, Samis EK, Lougheed SC (2008) Genetic variation across species' geographical ranges: the central-marginal hypothesis and beyond. Mol Ecol 17:1170–1188
Erb LP, Ray C, Guralnick R (2011) On the generality of a climate-mediated shift in the distribution of the American pika (Ochotona princeps). Ecology 92:1730–1735
Excoffier L, Foll M, Petit RJ (2009) Genetic consequences of range expansions. Annu Rev Ecol Evol Syst 40:481–501
Flanagan SP, Forester BR, Latch EK, Aitken SN, Hoban S (2018) Guidelines for planning genomic assessment and monitoring of locally adaptive variation to inform species conservation. Evol Appl 11:1035–1052
Foll M, Gaggiotti O (2008) A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180:977–993
Franks SJ, Hoffmann AA (2012) Genetics of climate change adaptation. Annu Rev Genet 46:185–208
Frichot E, François O (2015) LEA: an R package for landscape and ecological association studies. Methods Ecol Evol 6:925–929
Frichot E, Schoville SD, Bouchard G, François O (2013) Testing for associations between loci and environmental gradients using latent factor mixed models. Mol Biol Evol 30:1687–1699
Funk WC, McKay JK, Hohenlohe PA, Allendorf FW (2012) Harnessing genomics for delineating conservation units. Trends Ecol Evol 27:489–496
Galbreath KE, Hafner DJ, Zamudio KR (2009) When cold is better: climate-driven elevation shifts yield complex patterns of diversification and demography in an Alpine specialist (American Pika, Ochotona Princeps). Evolution 63:2848–2863
Gautier M (2015) Genome-wide scan for adaptive divergence and association with population-specific covariates. Genetics 201:1555–1579
Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ et al. (2008) High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36:3420–3435
Gradogna A, Gavazzo P, Boccaccio A, Pusch M (2017) Subunit‐dependent oxidative stress sensitivity of LRRC8 volume‐regulated anion channels. J Physiol 595:6719–6733
Hafner DJ, Sullivan RM (1995) Historical and ecological biogeography of Nearctic pikas (Lagomorpha: Ochotonidae). J Mammal 76:302–321
Hafner DJ, Smith AT (2010) Revision of the subspecies of the American pika, Ochotona princeps (Lagomorpha: Ochotonidae). J Mammal 91:401–417
Hanson JO, Marques A, Veríssimo A, Camacho-Sanchez M, Velo-Antón G, Martínez-Solano Í et al. (2020) Conservation planning for adaptive and neutral evolutionary processes. J Appl Ecol 57:2159–2169
Harrisson KA, Pavlova A, Telonis-Scott M, Sunnucks P (2014) Using genomics to characterize evolutionary potential for conservation of wild populations. Evol Appl 7:1008–1025
Heled J, Drummond AJ (2008) Bayesian inference of population size history from multiple loci. BMC Evol Biol 8:289
Henry P, Russello MA (2013) Adaptive divergence along environmental gradients in a climate-change-sensitive mammal. Ecol Evol 3:3906–3917
Henry P, Sim Z, Russello MA (2012) Genetic evidence for restricted dispersal along continuous altitudinal gradients in a climate change-sensitive mammal: the American pika. PLoS One 7:e39077
Hewitt G (2000) The genetic legacy of the Quaternary ice ages. Nature 405:907–913
Hewitt GM (1996) Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc 58:247–276
Hinzpeter A, Lipecka J, Brouillard F, Baudoin-Legros M, Dadlez M, Edelman A et al. (2006) Association between Hsp90 and the ClC-2 chloride channel upregulates channel function. Am J Physiol Cell Physiol 290:C45–56
Hoban S (2018) Integrative conservation genetics: Prioritizing populations using climate predictions, adaptive potential and habitat connectivity. Mol Ecol Resour 18:14–17
Hoffmann AA, Sgrò CM (2011) Climate change and evolutionary adaptation. Nature 470:479–485
Holbrook JD, DeYoung RW, Janecka JE, Tewes ME, Honeycutt RL, Young JH (2012) Genetic diversity, population structure, and movements of mountain lions (Puma concolor) in Texas. J Mammal 93:989–1000
IPCC (2014) AR5 Synthesis Report: Climate Change 2014. IPCC.
Jentsch TJ, Lutter D, Planells-Cases R, Ullrich F, Voss FK (2016) VRAC: molecular identification as LRRC8 heteromers with differential functions. Pflüg Arch Eur J Physiol 468:385–393
Johnston AN, Bruggeman JE, Beers AT, Beever EA, Christophersen RG, Ransom JI (2019) Ecological consequences of anomalies in atmospheric moisture and snowpack. Ecology 100:e02638
Johnston KM, Freund KA, Schmitz OJ (2012) Projected range shifting by montane mammals under climate change: implications for Cascadia’s National Parks. Ecosphere 3:art97
Kilham L (1958) Territorial behavior in pikas. J Mammal 39:307–307
Kimura M (1971) Theoretical foundation of population genetics at the molecular level. Theor Popul Biol 2:174–208
Klingler KB, Jahner JP, Parchman TL, Ray C, Peacock MM (2021) Genomic variation in the American pika: signatures of geographic isolation and implications for conservation. BMC Ecol Evol 21:2
Lambers JHR (2015) Extinction risks from climate change. Science 348:501–502
Latch EK, Dharmarajan G, Glaubitz JC, Rhodes OE (2006) Relative performance of Bayesian clustering software for inferring population substructure and individual assignment at low levels of population differentiation. Conserv Genet 7:295–302
Latch EK, Scognamillo DG, Fike JA, Chamberlain MJ, Rhodes Jr OE (2008) Deciphering ecological barriers to North American River Otter (Lontra canadensis) gene flow in the Louisiana landscape. J Hered 99:265–274
Lee KM, Coop G (2017) Distinguishing among modes of convergent adaptation using population genomic data. Genetics 207:1591–1619
Lee KM, Coop G (2019) Population genomics perspectives on convergent adaptation. Philos Trans R Soc B Biol Sci 374:20180236
Lemay MA, Russello MA (2015) Genetic evidence for ecological divergence in kokanee salmon. Mol Ecol 24:798–811
Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv13033997 Q-Bio.
MacArthur RA, Wang LCH (1974) Behavioral thermoregulation in the pika Ochotona princeps: a field study using radiotelemetry. Can J Zool 52:353–358
McCain CM (2019) Assessing the risks to United States and Canadian mammals caused by climate change using a trait-mediated model. J Mammal 100:1808–1817
Meirmans PG, Van Tienderen PH (2004) GENOTYPE and GENODIVE: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Notes 4:792–794
Morin PA, Luikart G, Wayne RK, the SNP Workshop Group (2004) SNPs in ecology, evolution and conservation. Trends Ecol Evol 19:208–216
Moritz C, Agudo R (2013) The future of species under climate change: resilience or decline? Science 341:504–508
Moritz C, Patton JL, Conroy CJ, Parra JL, White GC, Beissinger SR (2008) Impact of a century of climate change on small-mammal communities in Yosemite National Park, USA. Science 322:261–264
Morrison SF, Hik DS (2007) Demographic analysis of a declining pika Ochotona collaris population: linking survival to broad-scale climate patterns via spring snowmelt patterns. J Anim Ecol 76:899–907
Moyer‐Horner L, Mathewson PD, Jones GM, Kearney MR, Porter WP (2015) Modeling behavioral thermoregulation in a climate change sentinel. Ecol Evol 5:5810–5822
Mussmann SM, Douglas MR, Chafin TK, Douglas ME (2019) BA3-SNPs: contemporary migration reconfigured in BayesAss for next-generation sequence data. Methods Ecol Evol 10:1808–1813
Parmesan C (2006) Ecological and evolutionary responses to recent climate change. Annu Rev Ecol Evol Syst 37:637–669
Paz-Vinas I, Loot G, Hermoso V, Veyssière C, Poulet N, Grenouillet G et al. (2018) Systematic conservation planning for intraspecific genetic diversity. Proc R Soc B Biol Sci 285:20172746
Peacock MM (1997) Determining natal dispersal patterns in a population of North American pikas (Ochotona princeps) using direct mark-resight and indirect genetic methods. Behav Ecol 8:340–350
Peacock MM, Smith AT (1997) Nonrandom mating in pikas Ochotona princeps: evidence for inbreeding between individuals of intermediate relatedness. Mol Ecol 6:801–811
Pew J, Muir PH, Wang J, Frasier TR (2015) related: an R package for analysing pairwise relatedness from codominant molecular markers. Mol Ecol Resour 15:557–561
Pickett STA (1989) Space-for-time substitution as an alternative to long-term studies. In: Likens GE (ed) Long-term studies in ecology. Springer New York, New York, NY, p 110–135
Piry S, Alapetite A, Cornuet J-M, Paetkau D, Baudouin L, Estoup A (2004) GENECLASS2: a software for genetic assignment and first-generation migrant detection. J Hered 95:536–539
R Core Team (2019) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, http://www.R-project.org/
Rankin AM, Galbreath KE, Teeter KC (2017) Signatures of adaptive molecular evolution in American pikas (Ochotona princeps). J Mammal 98:1156–1167
Rannala B, Mountain JL (1997) Detecting immigration by using multilocus genotypes. Proc Natl Acad Sci USA 94:9197–9201
Razgour O, Taggart JB, Manel S, Juste J, Ibáñez C, Rebelo H et al. (2018) An integrated framework to identify wildlife populations under threat from climate change. Mol Ecol Resour 18:18–31
Rellstab C, Gugerli F, Eckert AJ, Hancock AM, Holderegger R (2015) A practical guide to environmental association analysis in landscape genomics. Mol Ecol 24:4348–4370
Ritland K (1996) Estimators for pairwise relatedness and individual inbreeding coefficients. Genet Res 67:175–185
Robson KM, Lamb CT, Russello MA (2016) Low genetic diversity, restricted dispersal, and elevation-specific patterns of population decline in American pikas in an atypical environment. J Mammal 97:464–472
Rochette NC, Rivera‐Colón AG, Catchen JM (2019) Stacks 2: analytical methods for paired-end sequencing improve RADseq-based population genomics. Mol Ecol 28:4737–4754
Rousset F (2008) GENEPOP’007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8:103–106
Rubidge EM, Patton JL, Lim M, Burton AC, Brashares JS, Moritz C (2012) Climate-induced range contraction drives genetic erosion in an alpine mammal. Nat Clim Change 2:285–288
Russello MA, Waterhouse MD, Etter PD, Johnson EA (2015) From promise to practice: pairing non-invasive sampling with genomics in conservation. PeerJ 3:e1106
Savolainen O, Lascoux M, Merilä J (2013) Ecological genomics of local adaptation. Nat Rev Genet 14:807–820
Sjodin BMF, Galbreath KE, Lanier HC, Russello MA (2021) Chromosome-level reference genome assembly for the American pika (Ochotona princeps). J Hered https://doi.org/10.1093/jhered/esab031
Smith AT (1974b) The distribution and dispersal of pikas: influences of behavior and climate. Ecology 55:1368–1376
Smith AT (1974a) The distribution and dispersal of pikas: consequences of insular population structure. Ecology 55:1112–1119
Smith AT (2020) Conservation status of American pikas (Ochotona princeps). J Mammal 101:1466–1488
Smith AT, Ivins BL (1983) Colonization in a pika population: dispersal vs philopatry. Behav Ecol Sociobiol 13:37–47
Smith AT, Weston ML (1990) Ochotona princeps. Mamm Species 352:1–8.
Smith AT, Millar CI (2018) American pika (Ochotona princeps) population survival in winters with low or no snowpack. West North Am Nat 78:126–132
La Sorte FA, Jetz W (2010) Projected range contractions of montane biodiversity under global warming. Proc R Soc B Biol Sci 277:3401–3410
Stern DL (2013) The genetic causes of convergent evolution. Nat Rev Genet 14:751–764
Stewart JAE, Wright DH, Heckman KA (2017) Apparent climate-mediated loss and fragmentation of core habitat of the American pika in the Northern Sierra Nevada, California, USA. PLoS One 12:e0181834
Stewart JAE, Perrine JD, Nichols LB, Thorne JH, Millar CI, Goehring KE et al. (2015) Revisiting the past to foretell the future: summer temperature and habitat area predict pika extirpations in California. J Biogeogr 42:880–890
Varner J, Dearing MD (2014) The importance of biologically relevant microclimates in habitat suitability assessments. PLoS One 9:e104648
Waldvogel A-M, Feldmeyer B, Rolshausen G, Exposito‐Alonso M, Rellstab C, Kofler R et al. (2020) Evolutionary genomics can improve prediction of species’ responses to climate change. Evol Lett 4:4–18
Wang T, Hamann A, Spittlehouse DL, Murdock TQ (2012) ClimateWNA—high-resolution spatial climate data for Western North America. J Appl Meteorol Climatol 51:16–29
Waterhouse MD, Erb LP, Beever EA, Russello MA (2018) Adaptive population divergence and directional gene flow across steep elevational gradients in a climate-sensitive mammal. Mol Ecol 27:2512–2528
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wiens JJ (2016) Climate-related local extinctions are already widespread among plant and animal species. PLoS Biol 14:e2001104
Wilkening JL, Ray C (2016) Characterizing predictors of survival in the American pika (Ochotona princeps). J Mammal 97:1366–1375
Wilkening JL, Ray C, Beever EA, Brussard PF (2011) Modeling contemporary range retraction in Great Basin pikas (Ochotona princeps) using data on microclimate and microhabitat. Quat Int 235:77–88
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163:1177–1191
Wogan GOU, Wang IJ (2018) The value of space-for-time substitution for studying fine-scale microevolutionary processes. Ecography 41:1456–1468
Yandow LH, Chalfoun AD, Doak DF (2015) Climate tolerances and habitat requirements jointly shape the elevational distribution of the American pika (Ochotona princeps), with implications for climate change effects. PLoS One 10:e0131082
Zgurski JM, Hik DS (2012) Polygynandry and even-sexed dispersal in a population of collared pikas, Ochotona collaris. Anim Behav 83:1075–1082
Zhang D, Pan J, Cao J, Cao Y, Zhou H (2020) Screening of drought-resistance related genes and analysis of promising regulatory pathway in camel renal medulla. Genomics 112:2633–2639
Zhang Z, Schwartz S, Wagner L, Miller W (2000) A greedy algorithm for aligning DNA sequences. J Comput Biol 7:203–214
Acknowledgements
Computational resources were made available by Compute Canada through the Resources for Research Groups program. Funding for this work was provided by the Natural Sciences and Engineering Research Council of Canada (NSERC; Discovery Grant # RGPIN-2019-04621 to MAR). DAS was supported by a UBC International Doctoral Fellowship and BMFS was supported by an NSERC Postgraduate Scholarship.
Author information
Authors and Affiliations
Contributions
DAS participated in study design, conducted all laboratory work and data analyses, and drafted the manuscript. MWD participated in study design, collected field samples, and helped draft the manuscript. BMFS collected field samples and helped draft the manuscript. MAR conceived, designed, and coordinated the study, obtained funding, and helped draft the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by: Sam Banks
Supplementary information
Rights and permissions
About this article

Cite this article
Schmidt, D.A., Waterhouse, M.D., Sjodin, B.M.F. et al. Genome-wide analysis reveals associations between climate and regional patterns of adaptive divergence and dispersal in American pikas. Heredity 127, 443–454 (2021). https://doi.org/10.1038/s41437-021-00472-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-021-00472-3
