
Abstract
Genome sequences largely determine the biology and encode the history of an organism, and de novo assembly — the process of reconstructing the genome sequence of an organism from sequencing reads — has been a central problem in bioinformatics for four decades. Until recently, genomes were typically assembled into fragments of a few megabases at best, but now technological advances in long-read sequencing enable the near-complete assembly of each chromosome — also known as telomere-to-telomere assembly — for many organisms. Here, we review recent progress on assembly algorithms and protocols, with a focus on how to derive near-telomere-to-telomere assemblies. We also discuss the additional developments that will be required to resolve remaining assembly gaps and to assemble non-diploid genomes.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$189.00 per year
only $15.75 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others
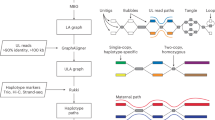

References
Schneider, V. A. et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 27, 849–864 (2017).
Jain, M. et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 36, 338–345 (2018).
C. elegans Sequencing Consortium. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282, 2012–2018 (1998).
Lander, E. S. et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001).
Myers, E. W. et al. A whole-genome assembly of Drosophila. Science 287, 2196–2204 (2000).
Venter, J. C. et al. The sequence of the human genome. Science 291, 1304–1351 (2001).
Bentley, D. R. et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456, 53–59 (2008).
Chin, C.-S. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569 (2013).
Koren, S. et al. Reducing assembly complexity of microbial genomes with single-molecule sequencing. Genome Biol. 14, R101 (2013).
Koren, S. et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 30, 693–700 (2012).
Koren, S. & Phillippy, A. M. One chromosome, one contig: complete microbial genomes from long-read sequencing and assembly. Curr. Opin. Microbiol. 23, 110–120 (2015).
Chaisson, M. J. P. et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature 517, 608–611 (2015).
Berlin, K. et al. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol. 33, 623–630 (2015).
Koren, S. et al. De novo assembly of haplotype-resolved genomes with trio binning. Nat. Biotechnol. 36, 1174–1182 (2018).
Wenger, A. M. et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 37, 1155–1162 (2019).
Jarvis, E. D. et al. Semi-automated assembly of high-quality diploid human reference genomes. Nature 611, 519–531 (2022). This work evaluates 23 developer-submitted assemblies of a diploid human sample and demonstrates the advantage of accurate long-read assembly.
Espinosa, E. et al. Comparing assembly strategies for third-generation sequencing technologies across different genomes. Genomics 115, 110700 (2023).
Gavrielatos, M., Kyriakidis, K., Spandidos, D. A. & Michalopoulos, I. Benchmarking of next and third generation sequencing technologies and their associated algorithms for de novo genome assembly. Mol. Med. Rep. 23, 251 (2021).
Chen, Y., Zhang, Y., Wang, A. Y., Gao, M. & Chong, Z. Accurate long-read de novo assembly evaluation with inspector. Genome Biol. 22, 312 (2021).
Eché, C. et al. A Bos taurus sequencing methods benchmark for assembly, haplotyping, and variant calling. Sci. Data 10, 369 (2023).
Nurk, S. et al. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 30, 1291–1305 (2020). This seminal paper reports the first T2T human genome.
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021). This paper describes hifiasm, a widely used assembler that produces high-quality assembly by integrating multiple data types.
Bankevich, A., Bzikadze, A. V., Kolmogorov, M., Antipov, D. & Pevzner, P. A. Multiplex de Bruijn graphs enable genome assembly from long, high-fidelity reads. Nat. Biotechnol. 40, 1075–1081 (2022). This paper describes the application of multiplex DBG to accurate long-read assembly.
Cheng, H. et al. Haplotype-resolved assembly of diploid genomes without parental data. Nat. Biotechnol. 40, 1332–1335 (2022).
Rautiainen, M. et al. Telomere-to-telomere assembly of diploid chromosomes with Verkko. Nat. Biotechnol. 41, 1474–1482 (2023). This paper describes Verkko, a tool that integrates PacBio HiFi and ONT ultra-long data for automated high-quality assembly.
Ekim, B., Berger, B. & Chikhi, R. Minimizer-space de Bruijn graphs: whole-genome assembly of long reads in minutes on a personal computer. Cell Syst. 12, 958–968.e6 (2021).
Cheng, H., Asri, M., Lucas, J., Koren, S. & Li, H. Scalable telomere-to-telomere assembly for diploid and polyploid genomes with double graph. Preprint at arXiv https://doi.org/10.48550/ARXIV.2306.03399 (2023).
Miga, K. H. et al. Centromere reference models for human chromosomes X and Y satellite arrays. Genome Res. 24, 697–707 (2014).
Stong, N. et al. Subtelomeric CTCF and cohesin binding site organization using improved subtelomere assemblies and a novel annotation pipeline. Genome Res. 24, 1039–1050 (2014).
Liao, W.-W. et al. A draft human pangenome reference. Nature 617, 312–324 (2023).
Gao, Y. et al. A pangenome reference of 36 Chinese populations. Nature 619, 112–121 (2023).
Rhie, A. et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature 592, 737–746 (2021). This paper presents 16 chromosomal assemblies of diverse vertebrate species, highlighting the improvements in assembly quality derived from long-read assembly.
Darwin Tree of Life Project Consortium. Sequence locally, think globally: the Darwin Tree of Life Project. Proc. Natl Acad. Sci. USA 119, e2115642118 (2022).
Lewin, H. A. et al. The Earth Biogenome Project 2020: starting the clock. Proc. Natl Acad. Sci. USA 119, e2115635118 (2022).
Smith, T. P. L. et al. The Bovine Pangenome Consortium: democratizing production and accessibility of genome assemblies for global cattle breeds and other bovine species. Genome Biol. 24, 139 (2023).
Nurk, S. et al. The complete sequence of a human genome. Science 376, 44–53 (2022).
Rhie, A. et al. The complete sequence of a human Y chromosome. Nature 621, 344–354 (2023).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Chin, C.-S. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054 (2016).
Shafin, K. et al. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat. Biotechnol. 38, 1044–1053 (2020).
Ruan, J. & Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 155–158 (2020).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Vaser, R. & Šikić, M. Time- and memory-efficient genome assembly with Raven. Nat. Comput. Sci. 1, 332–336 (2021).
Di Genova, A., Buena-Atienza, E., Ossowski, S. & Sagot, M.-F. Efficient hybrid de novo assembly of human genomes with WENGAN. Nat. Biotechnol. 39, 422–430 (2021).
Chin, C.-S. & Khalak, A. Human genome assembly in 100 minutes. Preprint at bioRxiv https://doi.org/10.1101/705616 (2019).
Xiao, C.-L. et al. MECAT: fast mapping, error correction, and de novo assembly for single-molecule sequencing reads. Nat. Methods 14, 1072–1074 (2017).
Chen, Y. et al. Efficient assembly of nanopore reads via highly accurate and intact error correction. Nat. Commun. 12, 60 (2021).
Hu, J. et al. An efficient error correction and accurate assembly tool for noisy long reads. Preprint at bioRxiv https://doi.org/10.1101/2023.03.09.531669 (2023).
Li, H. Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32, 2103–2110 (2016).
Kamath, G. M., Shomorony, I., Xia, F., Courtade, T. A. & Tse, D. N. HINGE: long-read assembly achieves optimal repeat resolution. Genome Res. 27, 747–756 (2017).
Lin, Y. et al. Assembly of long error-prone reads using de Bruijn graphs. Proc. Natl Acad. Sci. USA 113, E8396–E8405 (2016).
Ebert, P. et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 372, eabf7117 (2021).
Selvaraj, S., R. Dixon, J., Bansal, V. & Ren, B. Whole-genome haplotype reconstruction using proximity-ligation and shotgun sequencing. Nat. Biotechnol. 31, 1111–1118 (2013).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125 (2013).
Kaplan, N. & Dekker, J. High-throughput genome scaffolding from in vivo DNA interaction frequency. Nat. Biotechnol. 31, 1143–1147 (2013).
Garg, S. et al. Chromosome-scale, haplotype-resolved assembly of human genomes. Nat. Biotechnol. 39, 309–312 (2021).
Deshpande, A. S. et al. Identifying synergistic high-order 3D chromatin conformations from genome-scale nanopore concatemer sequencing. Nat. Biotechnol. 40, 1488–1499 (2022).
Falconer, E. et al. DNA template strand sequencing of single-cells maps genomic rearrangements at high resolution. Nat. Methods 9, 1107–1112 (2012).
Porubsky, D. et al. Fully phased human genome assembly without parental data using single-cell strand sequencing and long reads. Nat. Biotechnol. 39, 302–308 (2021).
Malinsky, M., Simpson, J. T. & Durbin, R. trio-sga: facilitating de novo assembly of highly heterozygous genomes with parent-child trios. Preprint at bioRxiv https://doi.org/10.1101/051516 (2016).
Wang, O. et al. Efficient and unique cobarcoding of second-generation sequencing reads from long DNA molecules enabling cost-effective and accurate sequencing, haplotyping, and de novo assembly. Genome Res. 29, 798–808 (2019).
Chen, Z. et al. Ultralow-input single-tube linked-read library method enables short-read second-generation sequencing systems to routinely generate highly accurate and economical long-range sequencing information. Genome Res. 30, 898–909 (2020).
Meier, J. I. et al. Haplotype tagging reveals parallel formation of hybrid races in two butterfly species. Proc. Natl Acad. Sci. USA 118, e2015005118 (2021).
Lam, E. T. et al. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat. Biotechnol. 30, 771–776 (2012).
Makova, K. D. et al. The complete sequence and comparative analysis of ape sex chromosomes. Preprint at bioRxiv https://doi.org/10.1101/2023.11.30.569198 (2023).
Naish, M. et al. The genetic and epigenetic landscape of the Arabidopsis centromeres. Science 374, eabi7489 (2021).
Wang, B. et al. High-quality Arabidopsis thaliana genome assembly with nanopore and HiFi long reads. Genom. Proteom. Bioinform. 20, 4–13 (2022).
Altemose, N. et al. Complete genomic and epigenetic maps of human centromeres. Science 376, eabl4178 (2022).
Vollger, M. R. et al. Increased mutation and gene conversion within human segmental duplications. Nature 617, 325–334 (2023).
Li, H. et al. A synthetic-diploid benchmark for accurate variant-calling evaluation. Nat. Methods 15, 595–597 (2018).
Ko, B. J. et al. Widespread false gene gains caused by duplication errors in genome assemblies. Genome Biol. 23, 205 (2022).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinform. 19, 460 (2018).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898 (2020).
Das, A. K., Goswami, S., Lee, K. & Park, S.-J. A hybrid and scalable error correction algorithm for indel and substitution errors of long reads. BMC Genom. 20, 948 (2019).
Holley, G. et al. Ratatosk: hybrid error correction of long reads enables accurate variant calling and assembly. Genome Biol. 22, 28 (2021).
Au, K. F., Underwood, J. G., Lee, L. & Wong, W. H. Improving PacBio long read accuracy by short read alignment. PLoS ONE 7, e46679 (2012).
Salmela, L. & Rivals, E. LoRDEC: accurate and efficient long read error correction. Bioinformatics 30, 3506–3514 (2014).
Hackl, T., Hedrich, R., Schultz, J. & Förster, F. proovread: large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics 30, 3004–3011 (2014).
Madoui, M.-A. et al. Genome assembly using Nanopore-guided long and error-free DNA reads. BMC Genom. 16, 327 (2015).
Goodwin, S. et al. Oxford Nanopore sequencing, hybrid error correction, and de novo assembly of a eukaryotic genome. Genome Res. 25, 1750–1756 (2015).
Miclotte, G. et al. Jabba: hybrid error correction for long sequencing reads. Algorithms Mol. Biol. 11, 10 (2016).
Haghshenas, E., Hach, F., Sahinalp, S. C. & Chauve, C. CoLoRMap: correcting long reads by mapping short reads. Bioinformatics 32, i545–i551 (2016).
Salmela, L., Walve, R., Rivals, E. & Ukkonen, E. Accurate self-correction of errors in long reads using de Bruijn graphs. Bioinformatics 33, 799–806 (2017).
Bao, E. & Lan, L. HALC: high throughput algorithm for long read error correction. BMC Bioinform. 18, 204 (2017).
Bao, E., Xie, F., Song, C. & Song, D. FLAS: fast and high-throughput algorithm for PacBio long-read self-correction. Bioinformatics 35, 3953–3960 (2019).
Wang, J. R., Holt, J., McMillan, L. & Jones, C. D. FMLRC: hybrid long read error correction using an FM-index. BMC Bioinform. 19, 50 (2018).
Mak, Q. X. C., Wick, R. R., Holt, J. M. & Wang, J. R. Polishing de novo nanopore assemblies of bacteria and eukaryotes with FMLRC2. Mol. Biol. Evol. 40, msad048 (2023).
Morisse, P., Lecroq, T. & Lefebvre, A. Hybrid correction of highly noisy long reads using a variable-order de Bruijn graph. Bioinformatics 34, 4213–4222 (2018).
Firtina, C., Bar-Joseph, Z., Alkan, C. & Cicek, A. E. Hercules: a profile HMM-based hybrid error correction algorithm for long reads. Nucleic Acids Res. 46, e125 (2018).
Zhang, H., Jain, C. & Aluru, S. A comprehensive evaluation of long read error correction methods. BMC Genom. 21, 889 (2020).
Guo, Y., Feng, X. & Li, H. Evaluation of haplotype-aware long-read error correction with hifieval. Bioinformatics 39, btad631 (2023).
Myers, E. W. Toward simplifying and accurately formulating fragment assembly. J. Comput. Biol. 2, 275–290 (1995).
Myers, E. W. The fragment assembly string graph. Bioinformatics 21, ii79–ii85 (2005).
Idury, R. M. & Waterman, M. S. A new algorithm for DNA sequence assembly. J. Comput. Biol. 2, 291–306 (1995).
Pevzner, P. A., Tang, H. & Waterman, M. S. An Eulerian path approach to DNA fragment assembly. Proc. Natl Acad. Sci. USA 98, 9748–9753 (2001).
Cordaux, R. & Batzer, M. A. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 10, 691–703 (2009).
Vrček, L., Bresson, X., Laurent, T., Schmitz, M. & Šikić, M. Learning to untangle genome assembly with graph convolutional networks. Preprint at arXiv https://doi.org/10.48550/arXiv.2206.00668 (2022).
Chikhi, R., Limasset, A. & Medvedev, P. Compacting de Bruijn graphs from sequencing data quickly and in low memory. Bioinformatics 32, i201–i208 (2016).
Peng, Y., Leung, H. C. M., Yiu, S. M. & Chin, F. Y. L. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428 (2012).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Rautiainen, M. & Marschall, T. MBG: minimizer-based sparse de Bruijn Graph construction. Bioinformatics 37, 2476–2478 (2021).
Ye, C., Ma, Z. S., Cannon, C. H., Pop, M. & Yu, D. W. Exploiting sparseness in de novo genome assembly. BMC Bioinform. 13, S1 (2012).
Roberts, M., Hayes, W., Hunt, B. R., Mount, S. M. & Yorke, J. A. Reducing storage requirements for biological sequence comparison. Bioinformatics 20, 3363–3369 (2004).
Edgar, R. Syncmers are more sensitive than minimizers for selecting conserved k-mers in biological sequences. PeerJ 9, e10805 (2021).
Kille, B., Garrison, E., Treangen, T. J. & Phillippy, A. M. Minmers are a generalization of minimizers that enable unbiased local Jaccard estimation. Bioinformatics 39, btad512 (2023).
Benoit, G. et al. High-quality metagenome assembly from long accurate reads with metaMDBG. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-01983-6 (2024).
Rautiainen, M. & Marschall, T. GraphAligner: rapid and versatile sequence-to-graph alignment. Genome Biol. 21, 253 (2020).
Li, H., Feng, X. & Chu, C. The design and construction of reference pangenome graphs with minigraph. Genome Biol. 21, 265 (2020).
Lorig-Roach, R. et al. Phased nanopore assembly with Shasta and modular graph phasing with GFAse. Preprint at bioRxiv https://doi.org/10.1101/2023.02.21.529152 (2023).
Edge, P., Bafna, V. & Bansal, V. HapCUT2: robust and accurate haplotype assembly for diverse sequencing technologies. Genome Res. 27, 801–812 (2017).
Tourdot, R. W., Brunette, G. J., Pinto, R. A. & Zhang, C.-Z. Determination of complete chromosomal haplotypes by bulk DNA sequencing. Genome Biol. 22, 139 (2021).
Akbari, V. et al. Parent-of-origin detection and chromosome-scale haplotyping using long-read DNA methylation sequencing and Strand-seq. Cell Genom. 3, 100233 (2023).
Zeng, X. et al. Chromosome-level scaffolding of haplotype-resolved assemblies using Hi-C data without reference genomes. Preprint at bioRxiv https://doi.org/10.1101/2023.11.18.567668 (2023).
Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics 39, btac808 (2023). This paper describes the current state of the art Hi-C scaffolding method.
Garg, S. Towards routine chromosome-scale haplotype-resolved reconstruction in cancer genomics. Nat. Commun. 14, 1358 (2023).
Mc Cartney, A. M. et al. Chasing perfection: validation and polishing strategies for telomere-to-telomere genome assemblies. Nat. Methods 19, 687–695 (2022).
Formenti, G. et al. Merfin: improved variant filtering, assembly evaluation and polishing via k-mer validation. Nat. Methods 19, 696–704 (2022).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Zimin, A. V. & Salzberg, S. L. The genome polishing tool POLCA makes fast and accurate corrections in genome assemblies. PLoS Comput. Biol. 16, e1007981 (2020).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2020).
Simpson, J. T. et al. Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods 14, 407–410 (2017).
Vaser, R., Sović, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746 (2017).
Morisse, P., Marchet, C., Limasset, A., Lecroq, T. & Lefebvre, A. Scalable long read self-correction and assembly polishing with multiple sequence alignment. Sci. Rep. 11, 761 (2021).
Hu, J. et al. NextPolish2: a repeat-aware polishing tool for genomes assembled using HiFi long reads. Genom. Proteom. Bioinform. https://doi.org/10.1093/gpbjnl/qzad009 (2024).
Du, K. et al. The sterlet sturgeon genome sequence and the mechanisms of segmental rediploidization. Nat. Ecol. Evol. 4, 841–852 (2020).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Levy Karin, E., Mirdita, M. & Söding, J. MetaEuk-sensitive, high-throughput gene discovery, and annotation for large-scale eukaryotic metagenomics. Microbiome 8, 48 (2020).
Huang, N. & Li, H. compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics 39, btad595 (2023).
Li, H. Protein-to-genome alignment with miniprot. Bioinformatics 39, btad014 (2023).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Mapleson, D., Garcia Accinelli, G., Kettleborough, G., Wright, J. & Clavijo, B. J. KAT: a K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics 33, 574–576 (2017).
Ewing, B. & Green, P. Base-calling of automated sequencer traces using Phred. II. Error probabilities. Genome Res. 8, 186–194 (1998).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Jain, C. et al. Weighted minimizer sampling improves long read mapping. Bioinformatics 36, i111–i118 (2020).
Jain, C., Rhie, A., Hansen, N. F., Koren, S. & Phillippy, A. M. Long-read mapping to repetitive reference sequences using Winnowmap2. Nat. Methods 19, 705–710 (2022).
Mikheenko, A., Bzikadze, A. V., Gurevich, A., Miga, K. H. & Pevzner, P. A. TandemTools: mapping long reads and assessing/improving assembly quality in extra-long tandem repeats. Bioinformatics 36, i75–i83 (2020).
Bzikadze, A. V., Mikheenko, A. & Pevzner, P. A. Fast and accurate mapping of long reads to complete genome assemblies with VerityMap. Genome Res. 32, 2107–2118 (2022).
Mikheenko, A., Prjibelski, A., Saveliev, V., Antipov, D. & Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 34, i142–i150 (2018).
Hui, J., Shomorony, I., Ramchandran, K. & Courtade, T. A. Overlap-based genome assembly from variable-length reads. In 2016 IEEE International Symposium on Information Theory (ISIT) 1018–1022 (IEEE, 2016).
Jain, C. Coverage-preserving sparsification of overlap graphs for long-read assembly. Bioinformatics 39, btad124 (2023).
Kamath, S. S., Bindra, M., Pal, D. & Jain, C. Telomere-to-telomere assembly by preserving contained reads. Preprint at bioRxiv https://doi.org/10.1101/2023.11.07.565066 (2023).
Boucher, C., Bowe, A., Gagie, T., Puglisi, S. J. & Sadakane, K. Variable-order de Bruijn graphs. In 2015 Data Compression Conference 383–392 (IEEE, 2015).
Belazzougui, D., Gagie, T., Mäkinen, V., Previtali, M. & Puglisi, S. J. Bidirectional variable-order de Bruijn graphs. In LATIN 2016: Theoretical Informatics (eds Kranakis, E. et al.) 164–178 (Springer, 2016).
Díaz-Domínguez, D., Onodera, T., Puglisi, S. J. & Salmela, L. Genome assembly with variable order de Bruijn graphs. Preprint at bioRxiv https://doi.org/10.1101/2022.09.06.506758 (2022).
Ohno, S., Christian, L. C. & Stenius, C. Nucleolus-organizing microchromosomes of Gallus domesticus. Exp. Cell Res. 27, 612–614 (1962).
Smith, J. et al. Differences in gene density on chicken macrochromosomes and microchromosomes. Anim. Genet. 31, 96–103 (2000).
Allendorf, F. W. et al. Effects of crossovers between homeologs on inheritance and population genomics in polyploid-derived salmonid fishes. J. Hered. 106, 217–227 (2015).
Lawniczak, M. K. N. et al. Standards recommendations for the Earth BioGenome Project. Proc. Natl Acad. Sci. USA 119, e2115639118 (2022).
Porubsky, D. et al. Gaps and complex structurally variant loci in phased genome assemblies. Genome Res. 33, 496–510 (2023).
Tan, K.-T., Slevin, M. K., Meyerson, M. & Li, H. Identifying and correcting repeat-calling errors in nanopore sequencing of telomeres. Genome Biol. 23, 180 (2022).
Sun, H. et al. Chromosome-scale and haplotype-resolved genome assembly of a tetraploid potato cultivar. Nat. Genet. 54, 342–348 (2022).
Bao, Z. et al. Genome architecture and tetrasomic inheritance of autotetraploid potato. Mol. Plant 15, 1211–1226 (2022).
Kolmogorov, M. et al. metaFlye: scalable long-read metagenome assembly using repeat graphs. Nat. Methods 17, 1103–1110 (2020).
Feng, X., Cheng, H., Portik, D. & Li, H. Metagenome assembly of high-fidelity long reads with hifiasm-meta. Nat. Methods 19, 671–674 (2022).
Feng, X. & Li, H. Towards complete representation of bacterial contents in metagenomic samples. Preprint at arXiv https://doi.org/10.48550/arXiv.2210.00098 (2022).
Song, B., Buckler, E. S. & Stitzer, M. C. New whole-genome alignment tools are needed for tapping into plant diversity. Trends Plant Sci. 29, 355–369 (2024).
Scalzitti, N., Jeannin-Girardon, A., Collet, P., Poch, O. & Thompson, J. D. A benchmark study of ab initio gene prediction methods in diverse eukaryotic organisms. BMC Genom. 21, 293 (2020).
Gabriel, L. et al. BRAKER3: fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS and TSEBRA. Preprint at bioRxiv https://doi.org/10.1101/2023.06.10.544449 (2023).
Acknowledgements
We are grateful to H. Cheng and C. Zhou for their helpful comments on the manuscript.
Author information
Authors and Affiliations
Contributions
Both authors contributed to all aspects of the Review.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Genetics thanks Zechen Chong and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, H., Durbin, R. Genome assembly in the telomere-to-telomere era. Nat Rev Genet (2024). https://doi.org/10.1038/s41576-024-00718-w
Accepted:
Published:
DOI: https://doi.org/10.1038/s41576-024-00718-w
