
Abstract
Cas13a has been used to target RNA viruses in cell culture, but efficacy has not been demonstrated in animal models. In this study, we used messenger RNA (mRNA)-encoded Cas13a for mitigating influenza virus A and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection in mice and hamsters, respectively. We designed CRISPR RNAs (crRNAs) specific for PB1 and highly conserved regions of PB2 of influenza virus, and against the replicase and nucleocapsid genes of SARS-CoV-2, and selected the crRNAs that reduced viral RNA levels most efficiently in cell culture. We delivered polymer-formulated Cas13a mRNA and the validated guides to the respiratory tract using a nebulizer. In mice, Cas13a degraded influenza RNA in lung tissue efficiently when delivered after infection, whereas in hamsters, Cas13a delivery reduced SARS-CoV-2 replication and reduced symptoms. Our findings suggest that Cas13a-mediated targeting of pathogenic viruses can mitigate respiratory infections.
Similar content being viewed by others

Main
There are 219 species of viruses that are known to infect humans1, of which 214 are RNA viruses2. It is estimated that viral infections contribute to approximately 6.6% of global mortality3. This is especially concerning, given that there are approximately 90 drugs (from 1963 to 2016) to treat only nine viral species4. In addition, there are approved vaccines for only 15 viral species. Reassortment and antigenic shift and drift for influenza, as well as antibody-dependent enhancement for SARS and possibly SARS-CoV-2, pose challenges to vaccine development5,6. These factors likely contribute to epidemics and pandemics. Human health is thus under constant threat due to emerging and reemerging viral infections7. Outbreaks of Zika8, Ebola9, the current SARS-CoV-2 pandemic10 and the potential for future influenza pandemics11 warrant the development of new classes of antiviral drugs4. Current drug development is focused on small molecules and neutralizing antibodies, which require high doses or frequent re-dosing to obtain functional outcomes12,13. Thus, it is crucial to address the need for antivirals that are broad spectrum, flexible and effective across multiple viral species or strains.
The discovery of an RNA-targeting, class II, type VI CRISPR–Cas system in bacteria has engendered tremendous interest for potential applications. The Cas13a–crRNA complex activates when the target RNA (trans-activating RNA (trRNA)) complements with crRNA, and the Cas protein initiates RNA cleavage14,15, due to the higher eukaryote and prokaryote nucleotide-binding domain16. This property has been used to detect specific transcripts in mixtures of nucleic acids15. The specific RNase activity of Cas13a and Cas13d17 was also used to knock down endogenous genes, and, more recently, two groups have demonstrated the ability of Cas13b or Cas13d to degrade influenza RNA18,19. In both cases, transient transfection with plasmids or stable cell lines expressing Cas13 were used prophylactically against influenza RNA. In each case, Cas13 was expressed at least 24 h before infection, clearly demonstrating proof of principle.
In this study, we examined crucial steps toward using this approach as a treatment for respiratory viral infections. Given the advantages of transient expression for treating infections, we developed synthetic mRNA to express LbuCas13a both with and without a nuclear localization signal (NLS) sequence, as influenza RNA can be localized to both compartments at different times after infection, and both constructs were tested for function within in-tube assays. Why synthetic mRNA? Synthetic mRNA expression is transient and has very little chance of integrating, and innate immune responses can be mitigated through sequence design, modified nucleotides and purification20,21,22. In addition, given that Cas13a is a bacterial protein, it is possible that there might be immune responses to the protein itself, and this is more likely if it is expressed for long periods of time. Transient expression with minimal innate responses gives the highest probability of repeat dosing in vivo. Thus, this approach might be crucial for repeat dosing of Cas13a in infected individuals without immune clearance of treated cells due to Cas13a-mediated immune responses.
The mRNA-based approach was initially benchmarked by examining knockdown of endogenous genes, using previously published target regions10. In examining these genes, and expanding the number of controls used, it was found that not all knockdown of RNA was mediated by Cas13a. Many controls were needed to ensure that the knockdown was Cas13a mediated and that enzymatic action was occurring. Once it was determined which controls were important, guides against PB1 were screened via quantitative polymerase chain reaction (qPCR), given after infection, simulating a treatment strategy. Why polymerase genes? They are highly conserved, and, without polymerase protein, neither viral mRNA nor replication can occur10,23,24. A potent guide against PB1 mRNA was found using this approach, but guides with broader sequence homology across flu strains were sought. By first examining homology among 108 vaccine strains, a stretch of nucleotides in PB2 was found to overlap most H1N1 strains, including the H1N1 pandemic strain from 2009 and H3N2 strains used in previous vaccines. Vaccine strains are a benchmark for circulating strain variation over time and, thus, are good candidates. At least two potent guides against this sequence were found. Subsequent tests after infection included combining guides and time-course studies yielding potent knockdown of influenza RNA. We then asked whether this same approach could be used against SARS-CoV-2, the virus causing the current pandemic. We delivered the mRNA both before and after infection, with cytopathic effect (CPE) and RNA copy number used as the metrics for success, demonstrating clear mitigation of SARS-CoV-2.
Finally, we demonstrated that this approach has translational potential by delivering Cas13 mRNA and guides formulated with a PBAE-based polymer25 via nebulizer post-influenza infection, simulating treatment. At day 3 after infection, degradation of influenza RNA in the lung was evaluated, demonstrating robust knockdown. In addition, using the Syrian hamster model of SARS-CoV-2 infection26,27,28, Cas13 mRNA and guides were delivered using the same formulations as mentioned above, 20 h before infection, demonstrating significant differences in body weight over a 6-d period. Infections were performed within the same 24-h time frame as delivery due to the rapid progression of the virus in this species29, as in recent studies using antibodies and small molecules30,31,32. Given that the peak of expression is approximately 24 h after transfection, this is a fair test of Cas13-mediated mitigation of SARS-CoV-2 infection.
Overall, we have demonstrated the necessity of controls for designing crRNAs, the possibility of pan-influenza targeting of viral strains and the ability to target relevant and emergent respiratory pathogens, both in vitro and in vivo.
Results
mRNA expressed Cas13a cleaves RNA in the presence of both crRNA and trRNA
Using rabbit reticulocyte lysates, LbuCas13a and LbuCas13a-NLS (a variant with an NLS sequence) mRNAs were translated in vitro and used to assess the RNA cleavage activity of the expressed RNases in conjunction with a guide RNA (crRNAs) and trRNAs (Supplementary Fig. 1a); both variants contain a V5-epitope tag sequence. crRNA and trRNA were derived from a genome segment of influenza virus A (IAV) (Supplementary Table 1). The RNaseAlert substrate fluorescence was the output of RNA cleavage. Cas13a and Cas13a-NLS RNA cleavage occurred only in the presence of both crRNA and trRNA but not in the presence of crRNA alone or a non-targeted crRNA (NTCR). RNA cleavage generated fluorescence, which increased to its maximum during the initial 10- and 20-min period, respectively, and then gradually decreased over time, likely due to photobleaching. The trend of RNA cleavage was similar for both Cas13a and Cas13a-NLS (Supplementary Fig. 1b). The products of the reactions were analyzed via gel electrophoresis, to visualize and quantify the product of the RNA cleavage activity—that is, the ‘RNA smear’ at the bottom of the gel (Supplementary Fig. 2). Our results demonstrated that, overall, the designed mRNAs express functional Cas13 proteins. Upon transfection of mRNA in A549 cells, LbuCas13a localized to the cytosol, and LbuCas13a-NLS had an additional nuclear distribution (Supplementary Fig. 3). Because, during infection, IAV is found both in the nucleus, where it replicates, and in the cytosol (Supplementary Fig. 4) of cells, both RNAses were concurrently further investigated.
Endogenous gene knockdown via mRNA-based Cas13 expression
To validate the mRNA-based approach, A549 cells were transfected with mRNA expressing LbuCas13a, with and without an NLS, and guides targeting the endogenous genes PPIB, CXCR4 and KRAS. Target gene knockdown was evaluated at 24 h via qPCR (Supplementary Fig. 5a–c and Supplementary Table 2). To date, most studies have evaluated only targeted reduction of RNA relative to the reduction due to an NTCR. Here, in addition to the NTCR, two additional controls were added: an mRNA expressing a ‘dead’ or inactive Cas13a plus targeted guide and a green fluorescent protein (GFP)-encoding mRNA plus targeted guide. Each control yielded valuable information regarding how the guide was performing. The dead version gives insight into whether ‘binding-only’ events might affect RNA levels, whereas the GFP control adds additional information regarding whether the guide alone has knockdown effects. For PPIB, the controls, in general, acted as expected. The PPIB guides exhibited 57% and 53% knockdown via the cytosolic and NLS versions of LbuCas13a in 24 h, respectively (Supplementary Fig. 5a). For CXCR4, unfortunately, the guides themselves, which used a published spacer sequence, significantly knocked down the mRNA by over 90% (Supplementary Fig. 5b) without Cas13a expression by a currently unknown mechanism. This is not necessarily a problem per se, but this control group helps distinguish ‘binding’-related knockdown from enzymatic effects. This information is important for establishing the mode of action of this therapeutic approach.
For KRAS, the guides, instead, exhibited Cas13-mediated knockdown, 38% and 60%, via the cytosolic and NLS versions of LbuCas13a in 24 h, respectively (Supplementary Fig. 5c). The data resulting from the CXCR4 experiments were surprising but clearly demonstrated the need for the additional controls when screening guides to ensure Cas13-mediated knockdown.
For both PPIB and KRAS, the slight increase in gene expression observed in the NLS, dead Cas13 plus target guide and GFP plus target guide conditions is likely due to transfection, as GFP alone induced a similar increase (Supplementary Fig. 6). Because the controls did not significantly decrease RNA levels, knockdown was due only to the active enzyme.
To address the occurrence of collateral RNase activity or off-target effects, the PPIB and KRAS knockdown experiments were repeated, and total RNA was extracted and processed for qPCR. The expression of 11 housekeeping genes and 18S rRNA were quantified by comparing the effect of the targeted guide to the NTCR. We observed no significant increase or decrease (fold change threshold = 2 and P value threshold P = 0.05) of gene expression with respect to the control condition (Supplementary Fig. 7) for either Cas13a or Cas13a-NLS.
Screening of crRNAs targeting PB1 after infection
Next, five PB1 guides targeting both the viral genome and mRNA were designed and screened after infection (Supplementary Table 2). Here, A549 cells were first infected with influenza A/WSN/33 at a multiplicity of infection (MOI) of 0.01; 24 h after infection, mRNA and guides were delivered, and, at 48 h after infection, PB1 RNA levels were evaluated by qPCR. From this initial screen, one mRNA targeted guide (WSN33_PB1_m5) exhibited very strong knockdown, approximately 83% using the cytosolic Cas13a and 78% via the NLS version, during the 24-h period (Fig. 1a and Supplementary Fig. 8). None of the controls demonstrated ‘guide-only’ effects (Fig. 1b,c and Supplementary Fig. 8), and even the dead version showed minimal knockdown, demonstrating that the effects were likely only from enzymatic action.

a, Schematic representation of A/WSN/33 genome showing the localization of the crRNA guides. b, A549 cells were infected with IAV A/WSN/33 at MOI = 0.01. Twenty-four hpi, cells were transfected with Cas13a or Cas13a-NLS and 1:50 mol ratio of WSN33_PB1 or NTCR guides. Two-way ANOVA with Sidak’s multiple comparisons, where **P = 0.0001 and ****P < 0.0001. c, A549 cells were infected with A/WSN/33. Twenty-four hpi, cells were transfected with either Cas13a or Cas13a-NLS, dead Cas13a, dead Cas13a-NLS mRNA or an equal molar amount of GFP mRNA with a 1:50 mol ratio of WSN33_PB1_g2 or NTCR guides. Two-way ANOVA with Sidak’s multiple comparisons, where no significant differences were found. d, A549 cells were infected with A/WSN/33. Twenty-four hpi, cells were transfected with either Cas13a or Cas13a-NLS, deadCas13a, dead Cas13a-NLS mRNA or an equal molar amount of GFP mRNA with a 1:50 mol ratio of WSN33_PB1_m5 or NTCR guides. Two-way ANOVA with Sidak’s multiple comparisons, where ***P = 0.0006 and ****P < 0.0001. Means and standard deviations are shown in gray, with n = 6 biological replicates. WSN/33_PB1 gene copy numbers were normalized by NTCR values. NS, not significant.
Expanding the breath of guides against influenza: targeting PB2
Clearly, for this approach to have high therapeutic value, the ability to target many strains of a given virus is advantageous. For influenza, a bioinformatic comparison of 108 influenza historical vaccine strains and candidate vaccine strains, with publicly available genome sequences, yielded several conserved regions across different subsets of strains (Fig. 2). We specifically focused on our so-called ‘Human’ subset, which included human seasonal H1N1, H1N1pdm09 and human seasonal H3N2 strains. From this analysis, a conserved region in PB2 was found, and six guides were designed against both viral mRNA and the genome of influenza (Supplementary Table 2). Guides with a stable stem-loop structure and targeting regions without substantial base pairing were selected using computational folding prediction tools, resulting in consistent ΔG = −2.74 for the whole sequence and a positive ΔG for the target sequence (Fig. 2d and Supplementary Table 3). Three guides were found to knockdown the viral RNA > 50%, delivered 24 h after infection and evaluated at 48 h after infection, with varying effectiveness in the nucleus and cytosol (Fig. 3 and Supplementary Fig. 9). Guides WSN33_PB2_m4 and WSN33_PB2_g2 both exhibited over 75% knockdown at the RNA level (Fig. 3 and Supplementary Fig. 9). These two guides were examined for breadth of protection, and, when the combination was compared with over 52,000 strains, over the last 100 years, they were exact matches with 99.1% of strains (Fig. 2e,f and Supplementary Table 4). This result was very encouraging toward the goal of a pan-influenza approach.

a, Schematic overview of PB2 guide selection, validation, screening and coverage analysis. b, Breakdown of IAV candidate subtypes used for consensus searches. H1N1, human seasonal H1N1 and H1N1pdm09 vaccine strains; H3N2, human seasonal H3N2 vaccine strains; Swine, swine-origin H1Nx(v) and H3N2 (v) vaccine candidates; H5Nx, avian and avian-origin H5N1, H5N6 and H5N8 vaccine candidates; H7Nx, avian and avian-origin H7N9 and H7N7 vaccine candidates; H9N2, avian and avian-origin H9N2 vaccine candidates. c, Schematic representation of A/WSN/33 genome showing the localization of the crRNA guides with consensus regions alignments between the H1N1 and H3N2 subtypes. Guides were tiled by single-nucleotide shifts along the conserved region. d, Guides (with the Cas13a-Lbu header region) were folded and accepted when no folds exist beyond the initial direct-repeat stem-loop in the header region. e, After in vitro screening, WSN33_PB2_g2 and WSN33_PB2_m4 were aligned across 52,074 influenza A sequences since 1918. f, Percent coverage of both WSN33_PB2_g2 and WSN33_PB2_m4 across H1N1, H3N2 and H2N2 sequences. Gray region indicates years in which sequence data were available.

a, A549 cells were infected with A/WSN/33. Cells were transfected with Cas13a or Cas13a-NLS and 1:50 mol ratio of WSN33_PB2 or NTCR guides Two-way ANOVA with Sidak’s multiple comparisons, where *P = 0.025, **P = 0.0082, ***P = 0.0005 and ****P < 0.0001. b, A549 cells were infected with A/WSN/33. Twenty-four hpi, cells were transfected with Cas13a or Cas13a-NLS, dead Cas13a, dead Cas13a-NLS mRNA or an equal molar amount of GFP mRNA with a 1:50 mol ratio of WSN33_PB2_g2 or NTCR guides. Two-way ANOVA with Sidak’s multiple comparisons, where *P = 0.0199, **P = 0.0024, ***P = 0.0009 and ****P < 0.0001. c, A549 cells were infected with A/WSN/33. Twenty-four hpi, cells were transfected with Cas13a or Cas13a-NLS, dead Cas13a, dead Cas13a-NLS mRNA or an equal molar amount of GFP mRNA with a 1:50 mol ratio of WSN33_PB2_m4 or NTCR guide. Two-way ANOVA with Sidak’s multiple comparisons, where *P = 0.02, **P = 0.0048 and ***P = 0.0005. Means and standard deviations are shown in gray, with n = 6 biological replicates. WSN/33_PB2 gene copy numbers were normalized by NTCR values. NS, not significant.
Screening of crRNAs targeting PB2 after infection and in combination with a PB1 guide
The mRNA-targeted guide for PB1 was combined with both cytoplasmic and NLS-targeted Cas13a, and the combination was evaluated against a previously published small interfering RNA (siRNA)33. The use of the combination NLS and cytoplasmic Cas13a is justified by the variability of the distribution of viral ribonucleoproteins from cell to cell (Supplementary Fig. 4), and it yielded a ~15% increase in viral RNA knockdown (Supplementary Fig. 10). The mRNA-targeted guides for PB1 and PB2 were then combined with both cytoplasmic and NLS-targeted Cas13a, and the combination was evaluated both as a function of MOI and over time (Fig. 4). When the combination was evaluated in our standard screening assay, we clearly saw the benefits of the combination, yielding ~10% lower RNA values than with WSN33_PB1_m5 alone. When this combination was examined across MOI, it was clear that, at MOIs of 0.001 and 0.01, greater than 80% knockdown was achievable (Fig. 4a,b). The effect, though, might be increased if the enzymatic reaction is given more time and if the dose of mRNA and guide is increased. When the combination was evaluated over time at MOI = 0.01, the results demonstrated continued RNA reduction over a span of 72 h (Fig. 4c,d), with the peak difference from IAV and the NTCR at 48 h. It should be noted that we observed increased effects of the NTCR over time, likely due to low levels of NTCR overlap with native genes inducing activation. However, if the effect of the treatment or ‘drug’, Cas13a mRNA plus targeted guide, is compared with the IAV-only case, the approach was able to sustain 90% knockdown over the 3-d period.

a, A549 cells were infected with A/WSN/33 at MOI = 0.001, MOI = 0.01 or MOI = 0.1. Twenty-four hpi, cells were transfected with 0.25 µg of Cas13a, 0.25 µg of Cas13a-NLS, 1:50 mol ratio of WSN33_PB1_m5 and WSN33_PB1_m5/WSN33_PB2_m4 or NTCR guides. Twenty-four hpt (48 hpi), cells were lysed, and RNA was extracted. Kruskal–Wallis or Brown–Forsythe and Welch one-way ANOVA with Dunnett T3 multiple comparison tests, where * P = 0.0445, **P = 0.0098 and ***P = 0.008. b, A549 cells were infected with A/WSN/33 at MOI = 0.01. Twenty-four hpi, cells were transfected with Cas13a and Cas13a-NLS and 1:50 mol ratio of WSN33_PB1_m5 and WSN33_PB2_g2 or NTCR guides. Twenty-hour hpt (48 hpi), cells were lysed, and RNA was extracted. Brown–Forsythe and Welch one-way ANOVA with Dunnett T3 multiple comparison tests, where *P < 0.023 and **P < 0.0059. c, A549 cells were infected with A/WSN/33. Twenty-four hpi, cells were transfected with Cas13a and Cas13a-NLS with a 1:50 mol ratio of WSN33_PB1_m5/WSN33_PB2_m4 or NTCR guides. Twenty-four hpt (48 hpi), 48 hpt (72 hpi) or 72 hpt (96 hpi), cells were lysed, and RNA was extracted. Brown–Forsythe and Welch one-way ANOVA with Dunnett T3 multiple comparison tests were performed (left and middle), where *P = 0.0301 and **P = 0.0096. Two-way ANOVA with a Sidak’s multiple comparisons test (right) where **P = 0.0012 and ***P = 0.0005. Means and standard deviations are shown in gray, with n = 6 biological replicates. WSN/33_PB1 gene copy numbers were normalized by NTCR values. hpt, hours post treatment. NS, not significant.
Inhibition of SARS-CoV-2 in a Vero E6 model of infection
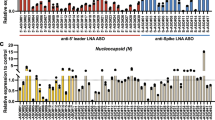
Given the importance of SARS-CoV-2, a set of crRNAs that target highly conserved regions in the replicase and nucleocapsid (N) regions of the genome were designed (Fig. 5 and Supplementary Tables 2, 3 and 5). The nucleocapsid sites would also allow for the targeting of any of the subgenomic RNAs produced by the virus. In this experiment, nine different guides were screened, by first transfecting Vero E6 cells overnight with the cytoplasmic version of Cas13a and guides and then infecting with MOI = ~0.1 of SARS-CoV-2. Note that only the cytoplasmic version was used, because the virus replicates in the cytosol34. Prophylactic delivery was initially used because this virus exhibits very rapid kinetics in Vero E6 cells, as well as the fact that they are interferon deficient. CPE was evaluated at 72 h after infection. From this screen, guides N3.2 (nucleocapsid), N3.1 (nucleocapsid), R5.1 (replicase) and N11.2 (nucleocapsid) were found to affect CPE, with guide N3.2 (Fig. 5b) exhibiting the lowest CPE. This was repeated using both individual and combinations of guides including all the controls and the data quantified by image analysis, demonstrating for the combination of N3.2 and N11.2 over 72% reduction in cell death, with over 80% of cells remaining in the plate (Fig. 5c). In addition, the experiment was repeated by first infecting Vero E6 with MOI = ~0.1 of SARS-CoV-2 and then transfecting cells either 1 hour post infection (hpi) or 4 hpi. The CPE assay demonstrated significant (>50%) reduction in cell death (Fig. 5d and Supplementary Fig. 11). Finally, the RNAse activity of Cas13 against SARS-CoV-2 was measured in both Vero E6 and Huh7 cells. Three hours after SARS-CoV-2 infection at an MOI of 2, cells were transfected with Cas13 mRNA along with either the N3.2 or NTCR guide RNA. qPCR, performed at 24 hpi, demonstrated significant knockdown of both the RNA-dependent RNA polymerase (RdRp) and N genes (Fig. 5e). Targeted Cas13a mRNA delivery resulted in 93.7% and 94.5% reduction in RdRp copy number in Vero E6 and Huh7 cells, respectively. Similarly, Cas13a mRNA delivery along with guide N3.2 reduced N copy number by 94.1% and 99.1% in Vero E6 and Huh7 cells, respectively.

a, Schematic representation of SARS-CoV and SARS-CoV-2 genomes showing the localization of the crRNA guides and selection process. Nineteen sequences of SARS-CoV-2 from the Wuhan region were aligned with the Toronto 2 SARS-CoV isolate sequence, and regions in ORF1ab and N with complete coverage were identified. Guide target sequences were tiled across the conserved region and checked for correct folding. b, Cas13a mRNA along with the indicated guides against N, ORF1ab (R5.1) or NTCR were transfected in Vero E6 cells overnight before infection with SARS-CoV-2 (MOI = 0.1). At 72 hpi, CPE was assessed, and percent live cell area was plotted. One-way ANOVA with multiple comparisons, where ****P < 0.0001, ***P = 0.0001 and **P = 0.0009. c, Cas13a mRNA along either with guide N3.2 alone or in the indicated combinations was transfected in Vero E6 cells overnight. GFP and dCas13a mRNA were used as controls. Cells were infected with SARS-CoV-2. At 72 hpi, CPE was assessed, and percent live cell area was plotted. One-way ANOVA with multiple comparisons where ****P < 0.0001, ***P = 0.0002 and **P = 0.0082. d, Vero E6 cells were infected with SARS-CoV-2. At 1 hpi, Cas13a mRNA along with guide N3.2 or NTCR were transfected. At 72 hpi, CPE was assessed, and percent live cell area was plotted. One-way ANOVA with multiple comparisons, where ****P = 0.0001 and ***P = 0.0003. Lines indicate means; error bars indicate s.d. (n = 3 independent biological replicates). e, Vero E6 or Huh7 cells were infected with SARS-CoV-2 (MOI = 2). At 3 hpi, Cas13a mRNA along with guide N3.2 or NTCR were transfected. At 24 hpi, qPCR was performed for RdRp or N copy number. Two-way ANOVA with multiple comparisons, where ****P < 0.0001 and *P = 0.0295. Lines indicate means; error bars indicate s.d. (n = 2 independent biological replicates).
Reduction of IAV in vivo after infection
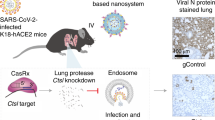
To test this approach in vivo, we developed both an apparatus and formulation for the RNA for nebulizer-based delivery. We designed a straightforward nose-cone nebulizer apparatus (Fig. 6a), which allows for up to three mice to be dosed simultaneously from a vibrating mesh nebulizer. PBAE was chosen for our formulation based on prior publication and lung delivery25. First, the final concentration of mRNA in the formulation was optimized using a glycosylphosphatidylinositol (GPI)-anchored nanoluciferase (aNLuc) encoding mRNA in the lung (Fig. 6b), yielding optimal expression in the lungs at 0.5 mg ml−1, as reported previously. Next, we determined whether influenza infection affects delivery. We infected mice with 3 lethal dose 50 (LD50) of influenza A/WSN/33 and then delivered aNLuc mRNA at 12 and 24 hpi as well as without infection (Fig. 6c,d). No significant differences in expression were observed, demonstrating efficient mRNA delivery and protein expression via nebulizer in the face of influenza infection. Finally, aNLuc mRNA was fluorescently labeled before delivery by nose-cone nebulization35,36. Four hours after delivery, mice were sacrificed, and lungs were extracted for IVIS imaging and fluorescence analysis of mRNA distribution within the organ. No significant differences in protein expression were measured between labeled and unlabeled mRNA (Supplementary Fig. 12). Confocal imaging revealed widespread distribution of labeled mRNA throughout the lungs (Supplementary Fig. 12).

a, Apparatus for mouse studies. b, 100 µg of aNLuc mRNA formulated was delivered to mice at the indicated final mRNA concentration. Lungs were analyzed at 1 d for luminescence as fold change of total flux relative to controls. Bars represent mean ± s.d. (n = 3). One-way ANOVA with multiple comparisons on log-transformed data, where ****P < 0.0001 and *P = 0.0211. c, Mice were infected with 3 LD50 A/WSN/33 as indicated after delivery of 100 µg of aNLuc mRNA. At 1 d after delivery, lungs were analyzed for luminescence. d, Quantification of part c as fold change total flux. Bars represent mean ± s.d. (n = 2). One-way ANOVA with multiple comparisons on log-transformed data, where ***P = 0.0005, **P = 0.001 and *P = 0.0011. e, Infected mice were dosed with Cas13a mRNA with guides at 6 hpi. Weights are plotted as mean ± s.e.m. (n = 5). f, Lung viral loads as mean fold change ± s.d. from the NTCR (n = 5). Unpaired two-tailed t-test on log-transformed data, where **P = 0.0063. g, Apparatus for hamster studies. h, Hamsters were dosed as indicated with aNLuc mRNA. Lungs were analyzed at 1 d for luminescence. i, Quantification of part h as fold change of total flux. Bars represent mean ± s.d. (n = 2). One-way ANOVA with multiple comparisons on log-transformed data, where ****P = 0.0002 and ***P = 0.0004. j, Hamsters were dosed with Cas13a mRNA with guides 20 h before infection with SARS-CoV-2 (n = 12 until day 3 and n = 6 until days 4–6). Lines indicate mean body weight normalized to day 0. Shaded region represents s.e.m. measured per cage. k, Lung viral loads from hamsters at 6 d after infection (n = 4). Data represent mean N copy number ± s.e.m.. Brown–Forsythe and Welch ANOVA with Dunnett’s multiple comparisons on log-transformed data, where **P = 0.0016 and *P = 0.0198.
To test this treatment approach, mice were infected with 3 LD50 of influenza A/WSN/33 via intranasal administration. Six hours after infection, one group was given, by nose-cone nebulizer, 100 µg of mRNA-encoding Cas13a (with and without NLS) and guide (WSN/33_PB1_m5) or an NTCR formulated with the PBAE-based polymer. Control groups included mice given 100 mg of Cas13a mRNA with an NTCR guide as well as an infection-only group. Mice body weight was recorded daily, and mice were euthanized 3 d after infection, when weight had dropped by ~15% in the NTCR condition (Fig. 6e). The viral RNA in mice lungs was quantified by qPCR. Analysis revealed an 89.1% (P = 0.0097) reduction of viral RNA from NTCR (Fig. 6f) and a 96.2% (P = 0.0001) from IAV only (Supplementary Fig. 13), demonstrating robust knockdown of viral RNA in vivo.
Mitigation of SARS-CoV-2 in vivo using the hamster model of infection
Once we observed Cas13 function and IAV RNA degradation in vivo, we then tested whether Cas13 could mitigate SARS-CoV-2 infections in the hamster model using body weight reduction prevention as a clinically relevant metric for success26,27,28. Given the size discrepancy between mice and Syrian hamsters, a modified nebulization apparatus was developed using larger-diameter animal restraints and 3D-printed nose cones but retaining the same nebulizer (Fig. 6g). Using a Cas13-nanoluciferase mRNA reporter, varying doses were tested, and 125 µg of mRNA was sufficient to induce robust expression (Fig. 6h,i). Next, 125 µg of mRNA-encoding Cas13a (without NLS) and guide (N3.2), formulated with the PBAE-based polymer, were delivered to the hamsters via the nose-cone nebulizer. Hamsters were infected with 103 plaque-forming units (PFU) of SARS-CoV-2 (USA-WA1/2020) via intranasal administration 20 h later. Control groups included mice given Cas13a mRNA with an NTCR guide, as well as an infection-only and mock group. Hamster body weights were recorded daily before euthanasia at 6 d after infection, and a clear difference was observed between the treated and untreated groups (Fig. 6j). We then used two different multiple linear regression models to analyze the weight data. First, we measured no statistical difference between the uninfected animals and the group that received the targeted guide (P = 0.111), demonstrating the ability of Cas13 to sufficiently alter SARS-CoV-2 pathogenesis in this model to abrogate weight loss (Supplementary Table 6). Both the NTCR (P = 0.0031) and infection-only (P = 0.0003) groups were statistically different from uninfected animals. Furthermore, we determined that the weight difference between the N3.2 and uninfected animals diverged quadratically with P = 0.00043 (Supplementary Table 7). In addition, hamster lung viral N RNA copy number was examined by qPCR at day 6 (Fig. 6k). We measured a significant decrease in SARS-CoV-2 copy number of 57% in the N3.2-treated animals relative to the NTCR and virus-only conditions.
Discussion
RNA viruses pose challenges for drug and vaccine development and are a global health concern. Unconventional molecular tools like RNA-activated RNases, such as Cas13a, could be a potential new paradigm for therapeutics against these pathogens. However, for the RNA targeting RNases to be safe and effective for therapeutic use, rapid, transient expression is preferred37. To achieve this, we opted for synthetic in vitro transcribed mRNA to express Cas13a. mRNA has the advantage of rapid translation of the desired protein, and it avoids safety concerns such as genome integration and unmitigated innate immune responses20,21,38,39,40.
Using this all-RNA-based approach, we found crRNA guides that targeted both the genome and the mRNA of influenza effectively, even when given 24 h after infection. To our knowledge, this is the first demonstration, to date, that this approach can be used after infection. There was a clear bias toward mRNA targeted guides, consistent with prior work using siRNA against influenza33. In addition, even though we were not able to find guides that were truly pan-influenza, they clearly overlap with over 52,000 strains, making significant strides toward this goal. Previous studies did not target polymerase genes. It is our contention that they are excellent targets owing to their clear role in generating viral RNA and the fact they are so well conserved. We also demonstrated how this approach scales with MOI and is functional over a 3-d period, even when using transient transfection of mRNA. The time-course data demonstrated the ability of Cas13a to degrade RNA at a rate equal to or more rapidly than the rate of RNA generation from viral replication. This has not been addressed to date. The data clearly show that, over the 3-d period after delivery, Cas13a kept RNA levels consistently ~1 log lower than without treatment. Future work will address how dose and Cas13 species affect these dynamics. A future therapeutic approach would most certainly contain multiple guides. We have demonstrated the use of two guides at once, but likely more guides will be used simultaneously, within the identified highly conserved regions, to prevent viral escape.
We then addressed the question: can this approach be applied directly to newly emerging pathogens such as SARS-CoV-2? From our results, it is clear that Cas13a can have significant effects on CPE, a well-established measure of antiviral activity41, or viral RNA levels when given before or after infection. These results clearly demonstrate the adaptability of this approach and its flexibility. Different guide sequences are sufficient to target either IAV or SARS-CoV-2. By delivering the nuclear or the cytoplasmic versions of Cas13, or both together, the replication and dynamics of different viruses can be significantly affected.
Finally, we answered a critical question regarding Cas13: can it be used in vivo against respiratory viruses? First, we formulated the mRNA with PBAE-based polymer and delivered it after infection using a nebulizer that is currently used by humans, simulating a treatment strategy, and demonstrated that Cas13 can degrade influenza viral RNA in vivo efficiently. Next, we assessed Cas13 effectiveness against SARS-CoV-2 infection in the Syrian hamster model and demonstrated that a single dose was sufficient to alter the pathophysiology of the infection, as indicated by the body weight and lung viral RNA data. Although we observed only a modest decrease in lung viral load, given the small sample size (n = 4), this is supported by the weak correlation between viral load and weight that was previously reported this model30. To our knowledge, this is the first demonstration of Cas13 mitigation, in vivo, of two important respiratory pathogens, in two different animal species. Our results bode well for addressing other infections. Future work will include more interrogations of delivery timing, as well as further development of mRNA carriers for the lung. Overall, this work demonstrates progress toward the use of this of Cas13 to treat respiratory viral infections.
Methods
Design and synthesis of Cas13a constructs and anchored nanoluciferase mRNA
The Cas13a sequence from Leptotrichia buccalis was obtained from Addgene (p2CT-His-MBP-Lbu_C2C2_WT, plasmid no. 83482). We cloned the wild-type Cas13a with a 3′ V5 tag (Cas13a-V5) and appended a 3′ untranslated region (UTR) from mouse alpha-globin (GenBank accession no. NM_001083955) in a pMA7 vector (GeneArt, Thermo Fisher Scientific). Additionally, we synthesized a construct with the Cas13a sequence from the L. buccalis along with an NLS sequence (at 3′ and 5′) and a 3′ V5 tag to create a Cas13a-V5-NLS version using GeneBlocks (Integrated DNA Technologies). For both constructs, we synthesized catalytically inactive versions (dCas13a-V5 and dCas13a-V5-NLS). The sequences were obtained fron Addgene (plasmid no. 100817). The sequences of the GPI anchor and nanoluciferase are described in Lindsay et al.35.
In vitro transcription of Cas13a
Plasmids were linearized with Not-I HF (New England Biolabs) overnight at 37 °C. Linearized templates were purified by sodium acetate (Thermo Fisher Scientific) precipitation and rehydrated with nuclease-free water. In vitro transcription was performed overnight at 37 °C using the HiScribe T7 Kit (NEB) following the manufacturer’s instructions (N1-methyl-pseudouridine modified). The resulting RNA was treated with DNase I (Aldevron) for 30 min to remove the template, and it was purified using lithium chloride precipitation (Thermo Fisher Scientific). The RNA was heat denatured at 65 °C for 10 min before capping with a Cap-1 structure using guanylyl transferase and 2′-O-methyltransferase (Aldevron). mRNA was then purified by lithium chloride precipitation, treated with alkaline phosphatase (NEB) and purified again. mRNA concentration was measured using a Nanodrop. mRNA stock concentrations were 1–3 mg ml−1. Purified mRNA products were analyzed by gel electrophoresis to ensure purity. crRNA guides were purchased from Integrated DNA Technologies or GenScript. The sequences are detailed in Supplementary Table 2.
RNA cleavage activity of in vitro translated of Cas13a
Cas13a mRNA was translated in vitro using the rabbit reticulocyte lysate system according to the manufacturer’s instructions (Promega). RNaseAlert-1 Substrate was mixed with crRNA or NTCR (500 nM) and trRNA (500 nM) in RNA processing buffer, consisting of A549 cells RNA (100 ng), 20 mM HEPES (pH 6.8), 50 mM KCl, 5 mM MgCl2, BSA (10 µg ml−1), yeast tRNA (10 µg ml−1), 0.01% Igepal CA-630 and 5% glycerol (Supplementary Table 1)15. This mixture was added cold to the translated Cas13a lysate (5 µl) in the 96-well plate wells and mixed well. All the reagent preparations and additions were performed on ice. The fluorescence measurements (excitation 485 ± 20 nm/emission 528 ± 20 nm) were recorded at room temperature for 90 min at 10-min intervals. The RNA cleavage products were run on a 15% TBE-urea gel. The gel was stained with SYBR Gold for 30 min and visualized under ultraviolet illumination. The gel was analyzed using Image Studio Lite quantification software (version 5.2, LI-COR) to quantify the intensity of the cleavage products (RNA smear).
Cell lines and viruses
All cell lines and viruses were purchased from the American Type Culture Collection (ATCC). Human lung epithelial cells A549 (CCL185), MDCK, Huh7 and Vero E6 were grown in media recommended by the ATCC. Guide screening experiments for IAV were performed in A549 cells. Guide screening experiments for SARS-CoV-2 were performed in Vero E6 cells. Influenza virus stocks (H1N1 influenza virus A/WSN/33) were prepared in MDCK cells. Briefly, MDCK cells were grown until 100% confluence in 175 mm2. The next day, cells were washed twice with PBS, and 1:1,000 dilution of virus was added in 5 ml of EMEM. Cells were then incubated with the virus for 1 h at room temperature on a rocker. Then, 25 ml of media was added to the cells. Cells were monitored for 72 h or until a severe CPE was observed. Virus was collected by centrifuging the cells at 1,000g for 10 min. Virus titers were determined by standard plaque assay.
SARS-CoV-2 (USA-WA1/2020) was obtained from BEI Resources. Viral stocks were generated by infecting Vero E6 cells (ATCC, C1008) at ~95% confluency in 150-cm2 flasks with SARS-CoV-2 at an MOI of 0.1 PFU per cell. At 68 h after infection, supernatants were collected, pooled and centrifuged at 400g for 10 min. The resulting stock was aliquoted, titered and stored at −80 °C for further use. All work with live SARS-CoV-2 was performed inside a certified Class II Biosafety Cabinet in a Biological Safety Level (BSL)-3 laboratory in compliance with all state and federal guidelines and with the approval of the University of Georgia Institutional Biosafety Committee.
Immunofluorescence assays
For all immunofluorescence assays, after the specified treatment, cells were fixed in 4% paraformaldehyde for 10 min, permeabilized with 0.02% Triton-X for 5 min and blocked with 5% BSA for 30 min. The primary antibodies used were the mouse monoclonal anti-influenza A H1N1, nucleoprotein (G20) (1:1,000, Kerafast EFD002) and the rabbit anti-V5 tag (1:250, Abcam, ab9116). The secondary antibodies used were Alexa Fluor 488 donkey anti-mouse (1:250, Thermo Fisher Scientific, A-21202) and Alexa Fluor 546 donkey anti-rabbit (1:250, Thermo Fisher Scientific, A10040). All antibodies were incubated for 30 min at 37 °C. Cells were routinely stained with DAPI before mounting with Prolong Gold (Thermo Fisher Scientific). Images were acquired with a Hamamatsu Flash 4.0 v2 sCMOS camera on a PerkinElmer UltraView spinning disk confocal microscope mounted to a Zeiss Axiovert 200 M body with a ×63 NA 1.4 plan-apochromat objective. Images were acquired with Volocity Acquisition Software (version 6.3.3, PerkinElmer) with z-stack intervals of 200 nm. Images were linearly contrast enhanced for visual clarity. Images were uniformly contrast enhanced across an experiment.
Optimization of mRNA transfection
A549 cells were seeded overnight at a density of 120,000–130,000 per well in a 24-well plate. The next day, cells were transfected with mRNA encoding for Cas13a variants and either targeted crRNA or non-targeted crRNA guides (NTCR) using Lipofectamine Messenger Max (Thermo Fisher Scientific) according to manufacturer instructions. Dead Cas13 constructs encoding mRNAs and GFP-encoding mRNA were also used as controls to assess the effect of Cas13a targeted binding and mRNA transfection/guides alone, respectively, on gene expression. For each well, 0.5 µg of mRNA and 1.5 µl of Messenger Max were used. crRNA guides and NTCR were added to the transfection mix at 20× molar guide:mRNA ratio in all endogenous gene knockdown experiments (PPIB, CXCR4 and KRAS). The sequences of the previously published crRNA guides10 are detailed in Supplementary Tables 2 and 3. For each well, 10 pmol siRNA and 1.5 µl of Messenger Max were used (Dharmacon; cat. no. PB1-2257, ON-TARGETplus Non-targeting siRNA #1).
Twenty-four hours after transfection, total RNA was extracted using the RNeasy Plus Mini Kit. Complementary DNA (cDNA) was prepared using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Thermo Fisher Scientific). qPCR experiments were performed using the FastAdvanced Master Mix (Thermo Fisher Scientific). Cas13-mediated degradation of PPIB, CXCR4 and KRAS mRNA was assayed by qPCR with the ΔΔCT method (n = 3) using GAPDH as control. Experiments were performed using a QuantStudio7 Flex thermal cycler (Applied Biosystems) using the QuantStudio7 software (version 1.3). All primer/probe assays for endogenous gene experiments were purchased from Thermo Fisher Scientific (Supplementary Table 8).
Evaluation of off-target effects
Cells were transfected with Cas13a or Cas13a-NLS and 20× molar excess of guides against PPIB or KRAS or NTCR as described. After 24 h, total RNA was extracted and quantified. cDNA was prepared using the RT2 First Strand Kit (Qiagen) according to manufacturer instructions. The expression of housekeeping genes was evaluated via qPCR using the SYBR Green-based commercially available RT2 housekeeping gene array (Qiagen) and analyzed using the RT2 Profiler PCR data. Analysis software is available online. The same cDNA samples were analyzed for 18S expression using the FastAdvanced Master Mix (Thermo Fisher Scientific) and Taqman primer/probe assays using GAPDH as an endogenous control (Supplementary Table 8).
In vitro antiviral assay with IAV
A549 cells were seeded overnight at a density of 120,000–130,000 per well in a 24-well plate. The next day, cells were washed with 1× PBS and infected by adding 100 µl of influenza virus diluted in serum-free media with TPCK for 1 h. The cells were then washed with 1× PBS, and complete media with TPCK were added for 24 h. Cells were then transfected with mRNA encoding for Cas13a and either targeted crRNA or non-targeted crRNA guides (NTCR) using Lipofectamine Messenger Max (Thermo Fisher Scientific) according to manufacturer instructions. Dead Cas13 constructs encoding mRNAs and GFP-encoding mRNA were also used as controls. For each well, 0.5 µg of mRNA and 1.5 µl of Messenger Max were used. crRNA guides and NTCR were added to the transfection mix at 50× molar guide:mRNA ratio in all IVA-based experiments. The sequences of the crRNA guides and ΔG values are detailed in Supplementary Tables 2 and 3. The sequences of the IAV strains used to design the guides against PB2 are available upon request from the Centers for Disease Control and Prevention.
Unless otherwise specified, 24 h after transfection, total RNA was extracted using RNeasy Plus Mini Kit. cDNA was prepared using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Thermo Fisher Scientific). qPCR experiments were performed using the FastAdvanced Master Mix (Thermo Fisher Scientific). The antiviral activity of Cas13a system was measured by qPCR (n = 6) with the absolute quantification of the viral PB1 or PB2 gene copy number. RT–qPCR technical triplicates were performed to determine biological replicate values. Experiments were performed using a QuantStudio7 Flex thermal cycler (Applied Biosystems).
In the ‘combo experiments’, for each well, 0.25 µg of each mRNA Cas13a and Cas13a-NLS and 1.5 µl of Messenger Max were used. Each crRNA guide was added to the transfection mix at 25× molar guide:mRNA ratio. The sequences of the primer/probe assays for PB1 and PB2 for H1N1 influenza virus A/WSN/33 are listed in Supplementary Table 8.
In vitro antiviral assay with SARS-CoV-2
Vero E6 cells were seeded overnight at ~80% confluency in a six-well plate. The next day, cells were transfected with mRNA encoding for Cas13a-V5 and either targeted crRNA or non-targeted crRNA guides (NTCR) using Lipofectamine Messenger Max (Thermo Fisher Scientific) according to manufacturer instructions. For each well, 5 µg of mRNA and 7.5 µl of Messenger Max were used. crRNA guides and NTCR were added to the transfection mix at 50× molar guide:mRNA ratio (Supplementary Tables 2 and 3). In the ‘combo experiments’, for each well, 5 µg of Cas13a and 7.5 µl of Messenger Max were used. Each crRNA guide was added to the transfection mix to maintain the 50× molar guide:mRNA ratio. After an overnight incubation at 37 °C and 5% CO2, plates were transferred to the BSL-3 for infection. The medium was removed, and cells were infected with SARS-CoV-2 for 45–60 min at an MOI of ~0.1 PFU per cell. After infection, cells were overlayed with 1× DMEM with 1% FBS containing 1.2% Avicel RC-581 and incubated for 72 h. The overlay was removed and cells were rinsed with 1× PBS and fixed/stained with a crystal violet solution containing 2% methanol and 4% formaldehyde for 10 min. In post-infection experiments, cells were infected with SARS-CoV-2 for 45–60 min at an MOI of ~0.1 PFU per cell. Either 1 h or 4 h after infection, cells were transfected with 5 µg of Cas13a and 7.5 µl of Messenger Max and incubated for 72 h before fixation and staining. Images for presentation were white balanced, and intensities for analysis were not modified for calculations. All images were brought into Photoshop, converted to grayscale and color inverted. A region of interest of the same size was then used for all images, and the sum intensity was calculated by Volocity. Intensities were normalized to a blank region of each image to account for image-to-image variation. Signal was then normalized to the mock condition to yield a percent live cell area.
Animal studies
Six- to 8-week-old female BALB/c mice (Jackson Laboratories) were maintained under pathogen-free conditions in individually ventilated and watered cages kept at negative pressure. Mice were kept in rooms on a 12-h light/dark cycle with ambient temperature between 22.8 and 23.9 °C with 30–40% relative humidity. Food was provided to mice ad libitum. Animals were acclimatized for at least 6 d before the beginning of experiments. Animals were randomly distributed among experimental groups. Researchers were blinded to animal group allocation during data acquisition. Animals were sacrificed by CO2 asphyxiation. Infected animals were handled and kept under BSL-2 conditions until euthanized.
Hamster delivery optimization experiments were performed at the Georgia Institute of Technology. Four-week-old male LVG Golden Syrian Hamsters (Charles River Laboratories) were maintained under pathogen-free conditions in individually ventilated and watered cages kept at negative pressure. Hamsters were kept in rooms on a 12-h light/dark cycle with ambient temperature between 21.1 and 22.8 °C with 35–50% relative humidity. Food was provided to hamsters ad libitum. Animals were acclimatized for at least 6 d before beginning experiments. Animals were randomly distributed among experimental groups. Researchers were blinded to animal group allocation during data acquisition. Animals were sacrificed by CO2 asphyxiation.
All animals were cared for according to the Georgia Institute of Technology Physiological Research Laboratory policies and under ethical guidance from the university’s Institutional Animal Care and Use Committee following National institutes of Health (NIH) guidelines.
Hamster infections were performed at the University of Georgia. Outbred male LVG Golden Syrian hamsters, 3–4 weeks of age, were obtained from Charles River Laboratories. Hamsters were housed inside an animal BSL-3 room in a HEPA-filtered cage/rack system and provided food and water ad libitum. Animals were randomly assigned to groups by animal care staff blinded as to study design and treatment. Hamsters were acclimatized before use. Animals were cared for according to the University of Georgia Animal Health Research Center policies and under ethical guidance from the university’s Institutional Animal Care and Use Committee following NIH guidelines.
Nebulizer-based mRNA deliveries to mice and hamsters
Mice were loaded into a custom-built nose-only exposure system constructed of a clear PVC tee and animal restraints (CODA Small Mouse Holder, Kent Scientific). These were connected using a custom 3D-printed nose cone (3D Printing Tech) made of a flexible TPU material. The nebulizer (Aeroneb, Kent Scientific) was then placed on the upward facing port of the tee. Doses were added dropwise to the nebulizer at a rate of 25 µl per mouse per droplet. After each individual droplet was nebulized, the clear tee was inspected until the vaporized dose had cleared (approximately 15–45 s per drop). Droplets were added until the desired dose per animal was achieved. After the vapor had cleared after the last droplet, the mice were removed from the restraints.
For delivery of mRNA to hamsters, the exposure system was modified with larger animal restraints (CODA Large Mouse Holder, Kent Scientific) and a larger 3D-printed nose cone to fit the larger restraints. To account for the increase in tidal volume in hamsters compared to mice, doses were added dropwise to the nebulizer at a rate of 62.5 µl per hamster per droplet.
mRNA was formulated for nebulizer-based delivery using a hyperbranched PBAE as previously described25 with minor modifications. Diacrylate and amine monomers were purchased from Sigma-Aldrich. To synthesize hyperbranched hDD90-118, acrylate:backbone amine:trifunctional amine monomers were reacted modifying the ratio at 1:0.59:0.40. Monomers were stirred in anhydrous dimethylformamide at a concentration of 150 mg ml−1 at 40 °C for 4 h and then 90 °C for 48 h. The mixtures were allowed to cool to 30 °C, and end cap amine was added at 1.0 molar equivalent relative to the acrylate and stirred for an additional 24 h. The polymers were purified by dropwise precipitation into cold anhydrous diethyl ether spiked with glacial acetic acid, vortexed and centrifuged at 1,250g for 2 min. The supernatant was discarded, and the polymer was washed twice more in fresh diethyl ether and dried under vacuum for 48 h. This step was repeated until the supernatant in the precipitation process became transparent. Polymers were stored at −20 °C.
Before delivery to animals, 100 mM of sodium acetate pH 5.0 was used to both solubilize the hyperbranched PBAE and dilute mRNA before mixing. The final concentration of the mRNA was 0.5 mg ml−1, and the PBAE was used at a 50× molar ratio to the mRNA. The tubes were incubated at room temperature for 10 min, and the particles were loaded into the nebulizer as described above.
After euthanasia, whole lungs were collected and rinsed with PBS. Lungs were then placed into a solution of Nano-Glo substrate solution (Promega) diluted 50-fold in PBS. Lungs were incubated for 5 min and then placed onto black paper and imaged with an IVIS Spectrum CT (PerkinElmer). Lung luminescence was then quantified using Living Image software (version 4.7.4, PerkinElmer).
mRNA distribution in mice lungs
To assess the distribution of aNLuc-encoding mRNA within mice lungs upon nebulization, four oligonucleotides (Biosearch Technologies) complementary to the mRNA’s 3′ UTR were designed with the following sequences: MT1 5′-thiol-XTTTTTXGCAAGCCCCGCAGAAGX-3′, MT2 5′-thiol-TXTTATTXAGAGAAGAAGGGCAXGG-3′, MT3 5′-thiol-XTTTTTXACCAAGAGGXACAGGXGC-3′ and MT4 5′-thiol-XTTTTTTCXACUCAGGCXUUAUXC-3′, where the boldface indicates 2′-O-methyl RNA and X indicates T(C6-amino) modifications. The oligos were labeled with Cy3B NHS-ester dye according to manufacturer instructions and were annealed to the anchored VHH-encoding mRNA as previously described36. The resulting fluorescently labeled mRNA was delivered to mice via nose-cone nebulization. Four hours after delivery, mice were euthanized, and lungs were isolated and processed for O.C.T. embedding. Tissue blocks in O.C.T. were immediately snap frozen in dry ice-cooled isopentane and sectioned using a cryostat in 10-μm-thick tissue sections. Tissue sections were used to evaluate the distribution of nebulized mRNA in the lungs. Lungs from untreated animals were used as controls. Images were acquired via confocal microscopy and processed as above.
Influenza viral load quantification from lung tissues
Mice were infected intranasally with 3 LD50 influenza virus using 20 µl per nostril after brief anaesthesia by isoflurane. Virus was diluted in DMEM without modifications to the desired PFU dose. After euthanasia, lungs were harvested into PBS before downstream assays.
Lungs were dissociated as described previously39. Briefly, lungs were weighed before dissociation using a GentleMACS in a C Tube using the ‘lung 2’ setting (Miltenyi). After centrifugation at room temperature for 5 min at 500g, pellets were lysed in RLT Plus buffer (Qiagen) and further homogenized using NAVY tubes in a Bullet Blender (Next Advance). Lysates were centrifuged for 15 min at 4 °C and maximum speed to clarify the supernatant. Total RNA was then extracted following a RNeasy Plus Mini Kit (Qiagen) per manufacturer instructions. cDNA synthesis and qPCR were then performed as detailed above with the absolute quantification of the viral PB1 gene copy number.
SARS-CoV-2 infections and RNA extraction
Hamsters were anesthetized by intraperitoneal injection with 100 mg kg−1 of ketamine/5 mg kg−1 of xylazine. After loss of toe pinch reflex, SARS-CoV-2 was administered to each via intranasal route in a volume of 50 µl. Animals were then administered reversal agent (atipamezole, 0.15 mg kg−1) and placed on a heating pad until able to right themselves. Body weights and clinical signs were checked and recorded daily. For sample collection, hamsters were anesthetized as described above and administered pentobarbital (100 mg kg−1) via intraperitoneal injection. After exsanguination and pneumothorax, tissues were collected aseptically for analyses.
Whole lungs from individual hamsters were placed in 2 ml of DMEM 1% FBS containing antibiotics/antimycotics (D1) in C tubes and homogenized using a GentleMACS machine at ‘lung 2’ setting (Miltenyi). After centrifugation for 10 min at 1,000g, supernatant was removed, and remaining homogenates were resuspended in Trizol for RNA extraction. Chloroform-based phase separation and RNA precipitation was then performed followed by two ethanol washes.
SARS-CoV-2 qPCR measurements
cDNA was synthesized from cellular RNA extracted from cells or hamster lung tissue samples using a specific primer 5′- TCCTTTTTAGGCTCTGTTGGTGGG-3′ complementary to the 3′ end of the viral genomic RNA using the iScript Select cDNA Synthesis Kit (Bio-Rad). qPCR was performed using either an RdRp region primer set (forward: 5′-GAATAGAGCTCGCACCGTAGCTGG-3′ and reverse: 5′-GCTAGCCACTAGACCTTGAGATGCAT-3′) or a nucleocapsid (N) region primer set (forward: 5′-GGCCAACAACAACAAGGCCAAACTGTC-3′ and reverse: 5′-GTCCTTTTTAGGCTCTGT TGGTGGG-3′) to amplify the cDNA using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad). A Quant Studio 3 system (Applied Biosystems) was used for qPCR. The cycling parameters were 95 °C for 4 min followed by 95 °C for 10 s, 56 °C for 45 s, 95 °C for 1 min, 55 °C for 1 min, 60 °C for 10 s and 95 °C for 10 s. Each sample was assayed in duplicate. The RNA genome copy number per liter was calculated using the average of the duplicate cycle threshold values and a standard curve constructed using viral RNA extracted from virions and quantified using nanodrop (NanoDrop 2000, version 1.6.198).
Statistical analyses
All experiments are represented as a mean of three or six biological replicates as indicated. Power analyses for group sizes were calculated using G*Power (version 3.1, University of Dusseldorf). Data were analyzed and plotted using GraphPad Prism 8. Statistical analyses were performed between groups using either ordinary one-way or two-way analysis of variance (ANOVA) as specified in individual figure captions. Hamster weight data were analyzed using a multiple linear regression model using regressors against weight data in Microsoft Excel, version 2009. Regression was performed with two different sets of regressors: 1) day, virus only, NTCR, N3.2, delivery flag (on the day of delivery) and infection flag (on the day of infection), all compared against the mock group; or 2) day, day2× (mock or NTCR), and infection flag (on all days after infection), all compared to the 3.2 and virus-only groups.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data generated or analyzed during this study are included in this published article and its Supplementary Information files. The accession code for the sequence used in this study is NM_001083955. Accession codes for the influenza and SARS-CoV-2 strains used in this study are available in Supplementary Tables 4 and 5. Source data are provided with this paper.
References
Woolhouse, M., Scott, F., Hudson, Z., Howey, R. & Chase-Topping, M. Human viruses: discovery and emergence. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 2864–2871 (2012).
Woolhouse, M. E. J. & Brierley, L. Epidemiological characteristics of human-infective RNA viruses. Sci. Data 5, 180017 (2018).
Lozano, R. et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the global burden of disease study 2010. Lancet 380, 2095–2128 (2012).
De Clercq, E. & Li, G. Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 29, 695–747 (2016).
Kimberlin, D. W. & Whitley, R. J. Antiviral resistance: mechanisms, clinical significance, and future implications. J. Antimicrob. Chemother. 37, 403–421 (1996).
Irwin, K. K., Renzette, N., Kowalik, T. F. & Jensen, J. D. Antiviral drug resistance as an adaptive process. Virus Evol. 2, vew014 (2016).
Marston, H. D., Folkers, G. K., Morens, D. M. & Fauci, A. S. Emerging viral diseases: confronting threats with new technologies. Sci. Transl. Med. 6, 253ps210 (2014).
Baud, D., Gubler, D. J., Schaub, B., Lanteri, M. C. & Musso, D. An update on Zika virus infection. Lancet 390, 2099–2109 (2017).
Gire, S. K. et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 345, 1369–1372 (2014).
Abudayyeh, O. O. et al. RNA targeting with CRISPR–Cas13. Nature 550, 280–284 (2017).
Neumann, G., Noda, T. & Kawaoka, Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459, 931–939 (2009).
Kamath, A. V. Translational pharmacokinetics and pharmacodynamics of monoclonal antibodies. Drug Discov. Today Technol. 21-22, 75–83 (2016).
Bai, S. et al. A guide to rational dosing of monoclonal antibodies. Clin. Pharmacokinet. 51, 119–135 (2012).
Abudayyeh, O. O. et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353, aaf5573 (2016).
East-Seletsky, A. et al. Two distinct RNase activities of CRISPR–C2c2 enable guide-RNA processing and RNA detection. Nature 538, 270–273 (2016).
Shmakov, S. et al. Discovery and functional characterization of diverse class 2 CRISPR–Cas systems. Mol Cell 60, 385–397 (2015).
Konermann, S. et al. Transcriptome engineering with RNA-targeting Type VI-D CRISPR effectors. Cell 173, 665–676 (2018).
Freije, C. A. et al. Programmable inhibition and detection of RNA viruses using Cas13. Mol. Cell 76, 826–837 (2019).
Abbott, T. R. et al. Development of CRISPR as an antiviral strategy to combat SARSCoV-2 and influenza. Cell 181, 865–876 (2020).
Andries, O. et al. N(1)-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J. Control. Release 217, 337–344 (2015).
Loomis, K. H. et al. In vitro transcribed mRNA vaccines with programmable stimulation of innate immunity. Bioconjug. Chem. 29, 3072–3083 (2018).
Baiersdorfer, M. et al. A facile method for the removal of dsRNA contaminant from in vitro-transcribed mRNA. Mol. Ther. Nucleic Acids 15, 26–35 (2019).
Gorman, O. T., Donis, R. O., Kawaoka, Y. & Webster, R. G. Evolution of influenza A virus PB2 genes: implications for evolution of the ribonucleoprotein complex and origin of human influenza A virus. J. Virol. 64, 4893–4902 (1990).
Patel, H. & Kukol, A. Evolutionary conservation of influenza A PB2 sequences reveals potential target sites for small molecule inhibitors. Virology 509, 112–120 (2017).
Patel, A. K. et al. Inhaled nanoformulated mRNA polyplexes for protein production in lung epithelium. Adv. Mater. 31, e1805116 (2019).
Imai, M. et al. Syrian hamsters as a small animal model for SARS-CoV-2 infection and countermeasure development. Proc. Natl Acad. Sci. USA 117, 16587–16595 (2020).
Sia, S. F. et al. Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature 583, 834–838 (2020).
Takayama, K. In vitro and animal models for SARS-CoV-2 research. Trends Pharmacol. Sci. 41, 513–517 (2020).
Brocato, R. L. et al. Disruption of adaptive immunity enhances disease in SARS-CoV-2-infected Syrian hamsters. J. Virol. 94, e01683-20 (2020).
Rogers, T. F. et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science 369, 956–963 (2020).
Yuan, S. F. et al. Metallodrug ranitidine bismuth citrate suppresses SARS-CoV-2 replication and relieves virus-associated pneumonia in Syrian hamsters. Nat. Microbiol. 5, 1439–1448 (2020).
Baum, A. et al. REGN-COV2 antibodies prevent and treat SARS-CoV-2 infection in rhesus macaques and hamsters. Science 370, 1110–1115 (2020).
Ge, Q. et al. RNA interference of influenza virus production by directly targeting mRNA for degradation and indirectly inhibiting all viral RNA transcription. Proc. Natl Acad. Sci. USA 100, 2718–2723 (2003).
Fields, B. N., Knipe, D. M. & Howley, P. M. Fields Virology 5th edn (Wolters Kluwer Health/Lippincott Williams & Wilkins, 2007).
Lindsay, K. E. et al. Aerosol delivery of synthetic mRNA to vaginal mucosa leads to durable expression of broadly neutralizing antibodies against HIV. Mol. Ther. 28, 805–819 (2020).
Lindsay, K. E. et al. Visualization of early events in mRNA vaccine delivery in non-human primates via PET–CT and near-infrared imaging. Nat. Biomed. Eng. 3, 371–380 (2019).
Kaczmarek, J. C., Kowalski, P. S. & Anderson, D. G. Advances in the delivery of RNA therapeutics: from concept to clinical reality. Genome Med. 9, 60 (2017).
Schalk, J. A. et al. Preclinical and clinical safety studies on DNA vaccines. Hum. Vaccin. 2, 45–53 (2006).
Tiwari, P. M. et al. Engineered mRNA-expressed antibodies prevent respiratory syncytial virus infection. Nat. Commun. 9, 3999 (2018).
Svitkin, Y. V. et al. N1-methyl-pseudouridine in mRNA enhances translation through eIF2α-dependent and independent mechanisms by increasing ribosome density. Nucleic Acids Res. 45, 6023–6036 (2017).
Schmidtke, M., Schnittler, U., Jahn, B., Dahse, H. & Stelzner, A. A rapid assay for evaluation of antiviral activity against coxsackie virus B3, influenza virus A, and herpes simplex virus type 1. J .Virol. Methods 95, 133–143 (2001).
Acknowledgements
The authors would like to acknowledge I. York from the Influenza Division, National Center for Immunization and Respiratory Diseases, Centers for Disease Control and Prevention, for providing the influenza and SARS-CoV-2 sequences used in this study. The authors would also like to acknowledge the veterinarians and staff at both the Physiological Research Laboratory at the Georgia Institute of Technology and the Animal Health Research Center at the University of Georgia. The following reagent was deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, National Institute of Allergy and Infectious Diseases, NIH: SARS-Related Coronavirus 2, Isolate USA-WA1/2020, NR-52281. This study was supported by the Defense Advanced Research Projects Agency, grant no. HR00111920008.
Author information
Authors and Affiliations
Contributions
E.L.B., S.B., C.Z. and P.J.S. developed screening assays. E.L.B. performed PB1 guide screens, guide combinations, MOI and time-course studies in vitro. D.V. developed nose-cone nebulizer setup and optimized polymer-based delivery, performed in vivo experiments and data analysis and designed SARS-CoV-2 guides and transfections. S.B. and P.T. designed Cas13 mRNA and guides, and P.T. performed PB2 screens. C.Z. performed endogenous gene knockdown experiments. L.R. synthesized the polymer and assisted with in vivo studies. N.B., R.H. and M.G.F. assisted with polymer production. J.B. assisted with delivery experiment. H.P. synthesized mRNA and assisted with in vivo studies. D.V., C.Z., J.H., F.M., J.M. and E.R.L. performed SARS-CoV-2 CPE assays. J.H., J.M. and E.R.L. performed SARS-CoV-2 in vivo studies. M.B. and H.S. performed PCR on SARS-CoV-2 studies. B.V. performed the statistical analyses on the hamster weight data. P.J.S. designed the experiments and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–13 and Supplementary Tables 1–3 and 6–8
Supplementary Table 4
Accession numbers and details of influenza strains
Supplementary Table 5
Accession numbers and details of SARS-CoV-2 strains
Source data
Source Data Fig. 1
Unprocessed RNA gels from Supplementary Fig. 2
Rights and permissions
About this article

Cite this article
Blanchard, E.L., Vanover, D., Bawage, S.S. et al. Treatment of influenza and SARS-CoV-2 infections via mRNA-encoded Cas13a in rodents. Nat Biotechnol 39, 717–726 (2021). https://doi.org/10.1038/s41587-021-00822-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41587-021-00822-w
This article is cited by
-
Transcript-specific induction of stop codon readthrough using a CRISPR-dCas13 system
EMBO Reports (2024)
-
dCas13-mediated translational repression for accurate gene silencing in mammalian cells
Nature Communications (2024)
-
CRISPR technologies for genome, epigenome and transcriptome editing
Nature Reviews Molecular Cell Biology (2024)
-
Nanotechnology of inhalable vaccines for enhancing mucosal immunity
Drug Delivery and Translational Research (2024)
-
The collateral activity of RfxCas13d can induce lethality in a RfxCas13d knock-in mouse model
Genome Biology (2023)
