
Abstract
The rate-limiting enzyme in the conversion of cholesterol into bile acids is cholesterol 7alpha-hydroxylase (CYP7A1). An A to C substitution 278 bp upstream in the promoter of the CYP7A1 gene was found to be associated with variations in serum lipid levels in normolipidaemic populations. In the present study, we investigated the involvement of this polymorphism in four different lipid disorders: hypertriglyceridaemia (HTG), combined hyperlipidaemia (CH), familial dysbetalipoproteinaemia (FD) and familial hypercholesterolaemia (FH). In a normolipidaemic male population, homozygous for the apoE3 isoform, an association was found between the AA genotype and higher levels of serum triglycerides (AA: +34%, P=0.036). In HTG patients, the AA genotype was associated with significantly higher concentrations of total cholesterol (+23%, P=0.005). There was a tendency towards increased levels of serum triglycerides (+39%, P=0.06), VLDL-triglycerides (+48%, P=0.053) and VLDL-cholesterol (+35%, P=0.059). No significant associations were found between serum lipid levels and the CYP7A1 polymorphism in patients with CH, FD and FH. Our results show that the A-278C polymorphism in the CYP7A1 gene has an effect on triglyceride levels in normolipidaemic males and on cholesterol levels in patients with hypertriglyceridaemia.
Similar content being viewed by others

Introduction
It is well known that variation in serum lipid and lipoprotein levels is associated with the development and progression of atherosclerotic cardiovascular disease. Epidemiological studies demonstrate that about 50% of the variations in serum total and low density lipoprotein (LDL) cholesterol are caused by genetic factors.1, 2, 3 Polymorphisms in genes encoding proteins involved in cholesterol metabolism can therefore be important determinants for these interindividual differences. In addition, variations in such genes might also contribute to (or modulate) the development of disorders in which the lipid metabolism is disturbed.
The liver plays a central role both in the regulation and maintenance of the whole body sterol balance. Conversion of cholesterol into bile acids in the liver, together with secretion of cholesterol into bile is quantitatively the major pathway for eliminating cholesterol from the body.4 Several studies indicate that variations in the biosynthetic pathway of bile acids have important phenotypic consequences for both cholesterol and lipid metabolism.5, 6, 7, 8, 9, 10 The first and rate-limiting enzyme in the breakdown of cholesterol is cholesterol 7alpha-hydroxylase (CYP7A1).4, 11 An A to C conversion 278 bp upstream in the promoter has been found in the CYP7A1 gene.12 This polymorphism has been related to variations in serum lipid levels; however, results seem inconsistent. A significant increase in LDL-cholesterol was observed in homozygous C carriers, both in men and women.12 In the Framingham Offspring Study,13 in which more than 2000 subjects were studied, increased LDL-cholesterol levels were found, only in men. Furthermore, women homozygous for the C-allele had significantly lower triglyceride levels than heterozygotes.13 In another study, performed by Hegele et al,14 the polymorphism was inconsistently associated with serum lipid levels in three normolipidaemic Canadian populations. So far, the influence of this polymorphism has only been studied in normolipidaemic populations.
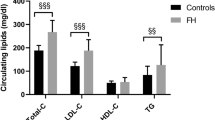
Several common inherited lipid disorders are described in humans; however, the precise molecular background of most of these disorders is yet unknown. Patients with familial dysbetalipoproteinaemia (FD or type III HLP) are characterised by elevated cholesterol and triglyceride levels due to the accumulation of chylomicron and very low-density lipoprotein (VLDL) remnants in the serum. Another common metabolic disorder is hypertriglyceridaemia (HTG), which is characterised by both overproduction and decreased clearance of triglyceride-enriched lipoproteins and by reduced concentrations of HDL-cholesterol. In combined hyperlipidaemia (CH), multiple lipoprotein phenotypes are present in the individual or within a family, mostly elevated serum LDL- and VLDL-cholesterol levels. Owing to the complex phenotypes of these diseases, they are thought to be heterogeneous multifactorial disorders, influenced by several genetic and environmental factors. In contrast, familial hypercholesterolaemia (FH) is a common monogenetic disorder caused by mutations in the LDL receptor gene, leading to elevated serum LDL-cholesterol concentrations, tendon xanthomas and premature development of coronary heart disease. Although the molecular mechanism underlying this defect is known, it is conceivable that additional genetic factors can contribute to the expression of the phenotype and thereby to the development and severity of this disorder.
Since these disorders are all characterised by disturbed serum lipid levels and CYP7A1 is a major regulator of sterol balance, but also may affect triglyceride levels,6 we investigated whether the CYP7A1 polymorphism is an additional genetic risk factor contributing to the expression or development of the four lipid disorders: HTG, FD, CH and FH.
Methods
Subjects
In this study, we used five different groups of subjects. The first group consisted of healthy normolipidaemic 35-year-old males (n=290), randomly selected from three different geographic areas in the Netherlands.15
Furthermore, we used four groups of hyperlipidaemic subjects. The first group were patients with endogenous HTG (n=139). The diagnosis HTG was based on the means of two fasting blood samples obtained after a dietary period of at least 8 weeks. The diagnostic criteria for HTG were: serum triglycerides >4.0 mmol/l, serum VLDL-cholesterol >1.0 mmol/l and serum LDL-cholesterol <4,5 mmol/l.16, 17
Patients with FD (n=157) were diagnosed on homozygosity for the apoE2 isoform.18 The FD population was divided into hypercholesterolaemic and normocholesterolaemic subjects. Hypercholesterolaemic FD patients were defined as having total cholesterol levels higher than the 90th percentile, whereas normocholesterolaemic E2/E2 subjects had total cholesterol levels lower than the 90th percentile, according to the age- and sex-related percentile levels of the Prospective Cardiovascular Munster Study.19
The diagnostic criteria for CH (n=92) were total serum cholesterol >7.5 mmol/l, serum triglyceride concentration >2 mmol/l, VLDL-cholesterol >1 mmol/l and absence of tendon xanthomas.18
Diagnosis of FH (n=272) was based on the mean of two measurements of total serum cholesterol >8.0 mmol/l, triglyceride levels <2.5 mmol/l (or a VLDL-cholesterol level of lower than 1.0 mmol/l after ultracentrifugation) and a family history of hypercholesterolemia and/or premature cardiovascular disease or the presence of tendon xanthomas.18
All serum lipid levels were measured before treatment with lipid-lowering drugs. Exclusion criteria for all disorders were secondary hyperlipidemia due to diabetes mellitus, renal, liver or thyroid disease, fasting glucose >7.0 mmol/l and alcohol consumption >40 g/day. Patients were not metabolically deranged.
All subjects gave informed consent and the study protocol was approved by the ethical committee from the Leiden University Medical Centre, Leiden, The Netherlands.
Biochemical analyses
Venous blood samples were collected after an overnight fast. Serum was obtained after centrifugation at 1500 g for 15 min at room temperature. Total serum cholesterol and triglyceride levels were measured enzymatically, using commercially available kits (Boehringer, Mannheim, Germany). For determination of individual lipoproteins, 3 ml of fresh serum was ultracentrifuged for 15 h at 232 000 g at 15°C in a TL-100 tabletop ultracentrifuge using a TLA-100.3 fixed angle rotor (Beckman, Palo Alto, CA, USA). The content of the ultracentrifuge tube was divided in a density (d) <1.006 and d=1.006–1.25 g ml−1 fraction, designated as the VLDL and LDL-HDL fraction, respectively. HDL-cholesterol was measured in the LDL-HDL fraction after precipitation of LDL with phosphotungstic acid and MgCl2. Insulin was measured with a conventional radioimmuno assay (Medgenix, Brussels, Belgium) and glucose with a Hitachi 747 analyzer, according to standard procedures (Roche diagnostics, Mannheim, Germany). In the control population, apoB concentrations were measured by immunonephelometric assay.20 and serum apoA1 levels were quantified by radial immunodiffusion.21 ApoE phenotyping in the normolipidaemic and the FD populations was performed using a rapid micro-method based on isoelectric focusing of delipidated plasma followed by immunoblotting using polyclonal anti-apoE antiserum.22 For the FD population, the results were confirmed by apoE genotyping as previously described by Reymer et al.23
CYP7A1 A-278C promoter polymorphism
Genomic DNA was isolated from peripheral blood leucocytes by standard methods.24 The A to C substitution 278 bp upstream of the translation initiation codon was detected as described previously;12 however, the following alternative primers were used: 5′-TTG AGG GAT GTT AGG TGA GTA-3′ (sense) and 5′-AAG AAT AAG CCA TAG ACA AC-3′ (antisense), resulting in a 690 bp fragment. Amplification was performed for 35 cycles of 30 s at 93°C, 30 s at 55°C and 30 s at 72°C with an initial denaturation period of 4 min. Reactions were performed in 25 μl volumes containing 14.3 μl milliQ water, 100–200 ng genomic DNA, 0.2 mM of each dNTP, 2.5 μl TAQ buffer, 0.5 U TAQ polymerase (Ht Biotechnology Ltd, UK) and 5 pmol of each primer. The PCR-amplified DNA fragment was digested by the enzyme BSAI, and fragments were resolved on a 2% agarose gel containing ethidium bromide.
Statistical analyses
For each population, the Hardy–Weinberg equilibrium was calculated by gene-counting and χ2 analysis. Genotype distributions between the different populations were tested with the χ2-test. Whenever serum lipid values showed no normal distribution, parameters were logarithmically transformed before analysis. Untransformed values are shown in the tables.
In the HTG and normolipidaemic FD populations, the number of women was small (n=19 and 18 respectively) and therefore man and women were combined. For the other populations, man and women were analysed separately; however, if there were no differences in results, the data were combined. Differences in serum lipid levels among the CYP7A1 genotypes were tested with analysis of variance. For the hyperlipidaemic populations, BMI, sex and age were taken as covariates. In case of significant differences, group means were compared by Fisher's Least Significant Difference test for multiple comparisons. Differences were considered significant at P<0.05.
Results
No dominant role of the CYP7A1 A-278C promoter polymorphism in hyperlipidaemic disorders
Baseline characteristics of the patient groups and normolipidaemic males are shown in Table 1. In comparison with the control group, either cholesterol, triglyceride or both levels were significantly elevated in the different patient groups, with the exception for the normolipidaemic FD patients.
All individuals were genotyped for the CYP7A1 A-278C polymorphism. The genotype distributions of the CYP7A1 variants, as well as the observed frequencies of alleles, among the different populations, are shown in Table 2. The observed allele frequencies in all populations were in Hardy–Weinberg equilibrium. There were no significant differences in genotype distributions between the normolipidaemic control population and the different hyperlipidaemic disorders. Since all FD patients carry the E2 isoform of the apoE protein but only 4% will develop the hyperlipidaemic phenotype, the FD population was divided into normolipidaemic and hyperlipidaemic subjects. However, no significant differences were found between genotype distributions of the normolipidaemic and the hyperlipidaemic FD patients. These data indicate that the CYP7A1 polymorphism plays no major role in the development of the four hyperlipidaemic disorders.
The CYP7A1 A-278C polymorphism has a modulating effect on lipid levels in normolipidaemic males and hypertriglyceridaemic patients
Since no data were available for LDL- and HDL-cholesterol in the normolipidaemic male population and apoB and apoA1 levels correlate well with LDL- and HDL-cholesterol levels, associations with these proteins (see Table 1) were determined. In the normolipidaemic male population, no significant associations were found between serum lipid and apolipoprotein levels and the CYP7A1 genotype. However, there was a tendency towards increased triglyceride levels in subjects with the genotype AA as compared to genotype CC (+12%, P=0.074) (data not shown). Since the apoE isoform is known to influence serum lipid levels,15 the analysis was repeated with subjects only having the E3E3 isoform, the most predominant isoform (49% of this control group). When only E3E3 isoforms were included in the analysis, a 34% increase (P=0.036) in serum triglycerides was found in individuals homozygous for the A allele as compared to individuals homozygous for the C allele (Figure 1). No associations were found between the polymorphism and serum total cholesterol, apoB and apoA1 levels.

Effect of the CYP7A1 genotype on serum triglyceride levels in a normolipidemic male population. Serum triglyceride levels according to CYP7A1 genotype are shown. Only carriers of the apoE3/E3 isoform were included in the analysis (n=143). AA, n=52; AC, n=72; CC, n=21.
To investigate the effect of the CYP7A1 polymorphism in the different lipid disorders, we divided the patients in homozygous A carriers and carriers of the C allele. The groups of the genotype CC were too small for statistical analysis.
HTG patients homozygous for the A allele had significantly increased levels of total cholesterol (P=0.006), total triglycerides (P=0.039), VLDL-cholesterol (P=0.05) and VLDL-triglycerides (P=0.039) (data not shown). The inclusion of BMI, sex and age in the model slightly changed these results, as shown in Figure 2. In subjects homozygous for the A allele, serum total cholesterol was significantly increased by 23% (P=0.005) as compared to carriers of the C allele. There was a strong tendency towards increased levels of serum triglycerides (+39% P=0.06), VLDL-triglycerides (48% P=0.053) and VLDL-cholesterol (+35%, P=0.059) in homozygous A carriers. No associations between the CYP7A1 genotype and serum HDL- and LDL-cholesterol levels were found in HTG patients. Since insulin strongly influences serum VLDL levels in the HTG patients,25 we compared serum insulin and glucose concentrations and insulin resistance (HOMA-index) between the homozygous A carriers and the carriers of the C polymorphism. However, no significant differences were found for these parameters between the groups (data not shown).

CYP7A1 genotype and effect on serum lipid levels in HTG patients. Serum total cholesterol (TC), total triglycerides (TG), VLDL-cholesterol (VLDL-C) and VLDL-triglycerides (VLDL-TG) in HTG patients according to homozygosity for the A allele (black bars, n=59) and homozygosity/heterozygosity for C carriers (white bars, n=80). Values are adjusted for BMI, sex and age.
In patients with CH and FH and both in normolipidaemic and hyperlipidaemic FD patients, no significant associations were found between the CYP7A1 genotype and serum levels of total cholesterol, total triglycerides, LDL-cholesterol and HDL-cholesterol. Furthermore, no differences in results were found between men and women in these groups (data not shown).
Discussion
In the present study we characterised the role of the A-278C promoter polymorphism in the cholesterol 7alpha-hydroxylase gene in a healthy normolipidaemic male population and in four groups of patients with lipid disorders: HTG, CH, FH and FD. Allele frequencies of all studied populations in our study are comparable to those in literature12; approximately 40% of the population carries the -278C allele. We did not find an effect on the frequency distribution of the A-278C polymorphism between the hyperlipidaemic patients and the normolipidaemic control population. This is a strong indicator that this variant does not contribute to the development of the hyperlipidaemic disorders. Detailed association studies, however, showed a modulating effect of the polymorphism on serum lipid levels.
In a healthy normolipidaemic male population, we found no significant associations between serum lipid levels and the CYP7A1 genotype. It is known, however, that the apolipoprotein E polymorphism is a modulator of serum cholesterol and triglyceride levels. Although results are variable, in several studies it was shown that the apoE polymorphism accounts for 2–20% of the interindividual differences in lipid and lipoprotein levels.26, 27, 28 For this reason we performed a second analysis in which we included only subjects homozygous for the E3 isoform, accounting for about 50% of the population. We found a significant 34% increase in serum total triglyceride levels in homozygous A carriers as compared to homozygous C carriers. Such a marked difference in triglyceride levels between homozygous A and C carriers has not been reported before, although Couture et al13 described a small difference between CC and AC carriers in women only. We found no association with apoB levels, in agreement with other reports.13, 14 An association of the CYP7A1 polymorphism has only been found with LDL-cholesterol levels, which were higher in carriers of the C allele,12 although this has not been confirmed consistently in other populations.13, 14 A reason for these differences may be differences in dietary intake and composition of the diet, which strongly affects serum lipid levels, and genetic background of the populations.
In HTG patients, we also found, for the first time, an association between the genotype AA and elevated levels of serum total cholesterol. Furthermore, there was a tendency towards increased levels of serum triglycerides, VLDL-triglycerides and VLDL-cholesterol.
Both in the healthy normolipidaemic male population and in the HTG population, the polymorphism seems to be involved in triglyceride metabolism. We did not find this association in the other hyperlipidaemic populations. For the FH population this association between the CYP7A1 polymorphism and plasma triglycerides was not expected. It is known that FH is caused by a defect in the LDL-receptor gene, leading to an increase in plasma LDL-cholesterol. This genetic defect does not lead to a change in plasma triglyceride levels. For the hyperlipidaemic FD population, an association between the CYP7A1 genotype and plasma triglycerides could be expected. These patients have high levels of triglycerides which could partly be explained by the CYP7A1 genotype. The same is true for the CH patients. This disorder is associated with a complex phenotype, and probably multiple genetic defects play a role. However, we found no evidence for a role of CYP7A1 in these diseases.
The molecular mechanism underlying the effect of the CYP7A1 polymorphism on serum lipid levels is as yet unknown. Studies on the transcriptional regulation of CYP7A1 revealed that the promoter region between nt −432 and −220 contains several cell-specific enhancer elements whose activity is controlled, in part, by HNF-3.29 It is conceivable, therefore, that the A-278C polymorphism might modulate transcriptional activity of the CYP7A1 gene and, consequently, the rate of cholesterol catabolism. Theoretically, the association that we found between the genotype AA and elevated triglyceride levels in normolipidaemic and HTG subjects could be explained by an increased bile acid synthesis in AA carriers. From previous studies it is known that there is a strong correlation between an increased bile acid synthesis and serum triglyceride levels.6 Treatment with the bile acid sequestrant cholestyramine, which induces bile acid synthesis, leads to an increase in VLDL-triglyceride and VLDL-cholesterol production. This increase is seen both in healthy individuals30 and in patients with various types of hyperlipidemia,31 and predominantly in patients with HTG.32, 33 In animals, this relation between bile acid synthesis and serum triglyceride levels also exists. Disruption of the enzyme sterol 27-hydroxylase in mice leads to a five-fold increase in CYP7A1 activity and a two-fold increase in hepatic and serum VLDL-triglyceride levels.5 For the HTG population this polymorphism could serve as an extra modulating genetic factor, increasing the triglycerides even more.
The hypothesis would also well be in line with previous research in which the genotype AA was found to be associated with decreased LDL-cholesterol levels in a normolipidaemic population.12 If the bile acid synthesis is upregulated, more cholesterol is needed and will be acquired by the liver via LDL-receptor mediated uptake and, as a consequence, LDL-cholesterol in serum will be decreased. However, again it should be noted that this hypothesis is only based on association studies, and therefore studies towards the functionality of the A-278C mutation are required. Furthermore, we cannot exclude that our findings are based on chance. Another possibility is that the polymorphism in itself is nonfunctional, and is in complete linkage disequilibrium with another, functional, polymorphism in the CYP7A1 gene or in another unidentified gene nearby the CYP7A1 locus.
In conclusion, these data show that the A-278C polymorphism in the CYP7A1 gene is associated with serum triglyceride levels in a normolipidaemic male population and with serum cholesterol levels in HTG patients. There are no associations between the CYP7A1 genotype and serum lipid levels in patients with CH, FD and FH.
References
Nora JJ, Berg K, Nora AH : Cardiovascular diseases. Genetics, epidemiology and prevention. New York: Oxford University Press, 1991.
Heller DA, de Faire U, Pedersen NL, Dahlen G, McClearn GE : Genetic and environmental influences on serum lipid levels in twins. N Engl J Med 1993; 328: 1150–1156.
Boomsma DI, Kempen HJ, Gevers Leuven JA, Havekes L, de Knijff P, Frants RR : Genetic analysis of sex and generation differences in plasma lipid, lipoprotein, and apolipoprotein levels in adolescent twins and their parents. Genet Epidemiol 1996; 13: 49–60.
Princen HMG, Post SM, Twisk J : Regulation of bile acid synthesis. Curr Pharmaceut Des 1997; 3: 59–84.
Repa JJ, Lund EG, Horton JD et al: Disruption of the sterol 27-hydroxylase gene in mice results in hepatomegaly and hypertriglyceridemia. Reversal by cholic acid feeding. J Biol Chem 2000; 275: 39685–39692.
The Lipid Research Clinics Coronary Primary Prevention Trial Results. II. The relationship of reduction in incidence of coronary heart disease to cholesterol lowering. JAMA 1984; 251: 365–374.
Poorman JA, Buck RA, Smith SA, Overturf ML, Loose-Mitchell DS : Bile acid excretion and cholesterol 7 alpha-hydroxylase expression in hypercholesterolemia-resistant rabbits. J Lipid Res 1993; 34: 1675–1685.
Kirk EA, Moe GL, Caldwell MT, Lernmark JA, Wilson DL, LeBoeuf RC : Hyper- and hypo-responsiveness to dietary fat and cholesterol among inbred mice: searching for level and variability genes. J Lipid Res 1995; 36: 1522–1532.
Machleder D, Ivandic B, Welch C, Castellani L, Reue K, Lusis AJ : Complex genetic control of HDL levels in mice in response to an atherogenic diet. Coordinate regulation of HDL levels and bile acid metabolism. J Clin Invest 1997; 99: 1406–1419.
Davis AM, Pond WG, Wheeler M et al: Alleles of the cholesterol 7 alpha-hydroxylase (CYP7) gene in pigs selected for high or low plasma total cholesterol. Proc Soc Exp Biol Med 1998; 217: 466–470.
Bertolotti M, Carulli N, Menozzi D et al: In vivo evaluation of cholesterol 7 alpha-hydroxylation in humans: effect of disease and drug treatment. J Lipid Res 1986; 27: 1278–1286.
Wang J, Freeman DJ, Grundy SM, Levine DM, Guerra R, Cohen JC : Linkage between cholesterol 7alpha-hydroxylase and high plasma low-density lipoprotein cholesterol concentrations. J Clin Invest 1998; 101: 1283–1291.
Couture P, Otvos JD, Cupples LA, Wilson PW, Schaefer EJ, Ordovas JM : Association of the A-204C polymorphism in the cholesterol 7alpha-hydroxylase gene with variations in plasma low density lipoprotein cholesterol levels in the framingham offspring study. J Lipid Res 1999; 40: 1883–1889.
Hegele RA, Wang J, Harris SB et al: Variable association between genetic variation in the CYP7 gene promoter and plasma lipoproteins in three Canadian populations. Atherosclerosis 2001; 154: 579–587.
Smit M, de Knijff P, Rosseneu M et al: Apolipoprotein E polymorphism in The Netherlands and its effect on plasma lipid and apolipoprotein levels. Hum Genet 1988; 80: 287–292.
Hau MF, Smelt AH, Bindels AJ et al: Effects of fish oil on oxidation resistance of VLDL in hypertriglyceridemic patients. Arterioscler Thromb Vasc Biol 1996; 16: 1197–1202.
Jonkers IJAM, Smelt AHM, Laarse van der A : Hypertriglyceridemia. Associated risks and effect on drug treatment. Am J Cardiovasc Drugs 2001; 1: 455–466.
Scriver CR, Beaudet AL, Sly WS, Valle D : The metabolic and molecular bases of inherited disease. Mc Graw-Hill, New York, 1995.
Assmann G, Schulte H : Results and conclusions of the prospective cardiovascular munster (PROCAM) Study; In: Assmann G (ed): Lipid metabolism disorders and coronary heart disease. Munich: MMV Medizin Verlag, 1993, pp 19–67.
Rosseneu M, Vinaimont N, Vercaemst R, Dekeersgieter W, Belpaire F : Standardization of immunoassays for the quantitation of plasma Apo B protein. Anal Biochem 1981; 116: 204–210.
Albers JJ, Wahl PW, Cabana VG, Hazzard WR, Hoover JJ : Quantitation of apolipoprotein A-I of human plasma high density lipoprotein. Metabolism 1976; 25: 633–644.
Havekes LM, de Knijff P, Beisiegel U, Havinga J, Smit M, Klasen E : A rapid micromethod for apolipoprotein E phenotyping directly in serum. J Lipid Res 1987; 28: 455–463.
Reymer PW, Groenemeyer BE, van de Burg R, Kastelein JJ : Apolipoprotein E genotyping on agarose gels. Clin Chem 1995; 41: 1046–1047.
Miller SA, Dykes DD, Polesky HF : A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215.
Asplund-Carlson A : Studies in Hypertriglyceridaemia, III: Glucose tolerance, insulin sensitivity and indices of adipose tissue lipolysis in randomly selected non-diabetic hypertriglyceridaemic swedish men. Eur J Clin Invest 1995; 25: 769–776.
Frikke-Schmidt R : Context-dependent and invariant associations between APOE genotype and levels of lipoproteins and risk of ischemic heart disease: a review. Scand J Clin Lab Invest Suppl 2000; 233: 3–25.
Haddy N, De Bacquer D, Chemaly M et al: The importance of plasma apolipoprotein E concentration in addition to its common polymorphism on inter-individual variation in lipid levels: results from Apo Europe. Eur J Hum Genet 2002; 10: 841–850.
Eichner JE, Dunn ST, Perveen G, Thompson DM, Stewart KE, Stroehla BC : Apolipoprotein E polymorphism and cardiovascular disease: a HuGE review. Am J Epidemiol 2002; 155: 487–495.
Molowa DT, Chen WS, Cimis GM, Tan CP : Transcriptional regulation of the human cholesterol 7 alpha-hydroxylase gene. Biochemistry 1992; 31: 2539–2544.
Carrella M, Ericsson S, Del Piano C, Angelin B, Einarsson K : Effect of cholestyramine treatment on biliary lipid secretion rates in normolipidaemic men. J Intern Med 1991; 229: 241–246.
Einarsson K, Hellstrom K, Kallner M : Bile acid kinetics in relation to sex, serum lipids, body weights, and gallbladder disease in patients with various types of hyperlipoproteinemia. J Clin Invest 1974; 54: 1301–1311.
Angelin B, Einarsson K, Hellstrom K, Leijd B : Bile acid kinetics in relation to endogenous tryglyceride metabolism in various types of hyperlipoproteinemia. J Lipid Res 1978; 19: 1004–1016.
Beil U, Crouse JR, Einarsson K, Grundy SM : Effects of interruption of the enterohepatic circulation of bile acids on the transport of very low density-lipoprotein triglycerides. Metabolism 1982; 31: 438–444.
Acknowledgements
We thank Wim van Duyvenvoorden for excellent technical assistance. This study was financially supported by a grant from ZonMw and the Netherlands Heart Foundation (NHS) (980-10-024).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hofman, M., Groenendijk, M., Verkuijlen, P. et al. Modulating effect of the A-278C promoter polymorphism in the cholesterol 7alpha-hydroxylase gene on serum lipid levels in normolipidaemic and hypertriglyceridaemic individuals. Eur J Hum Genet 12, 935–941 (2004). https://doi.org/10.1038/sj.ejhg.5201236
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201236
Keywords
This article is cited by
-
Bile diversion to the distal small intestine has comparable metabolic benefits to bariatric surgery
Nature Communications (2015)
-
CYP7A1 genotypes and haplotypes associated with hypertension in an obese Han Chinese population
Hypertension Research (2011)
-
Role of Cholesterol 7α-Hydroxylase (CYP7A1) in Nutrigenetics and Pharmacogenetics of Cholesterol Lowering
Molecular Diagnosis & Therapy (2006)
