
Abstract
The interferon regulatory factor 6 gene (IRF6) has been identified as the major Van der Woude (VWS) syndrome and popliteal pterygium (PPS) syndrome gene with mutations in the majority of the kindreds. We have studied altogether 17 kindreds from Sweden, Finland, Norway, Thailand and Singapore, and report here 10 mutations, six of them previously unseen. In two kindreds, we could document de novo mutations, both of them changing a codon for a glutamine residue to a stop. No mutation could be detected in the four VWS kindreds from Finland, suggesting a founder effect for a mutation in an atypical noncoding position. Our findings demonstrate that several distinct mutations occur in the Swedish population, and confirm the general notion of a broad spectrum of IRF6 mutations underlying the VWS/PPS phenotypes.
Similar content being viewed by others

Introduction
Van der Woude syndrome (VWS) is the most common oral cleft syndrome (MIM #119300). The genetically predisposed patients typically develop lip pits (LP), cleft lip (CL), cleft lip and palate (CLP), cleft palate (CP) and/or hypodontia (H).1 Popliteal pterygium syndrome (PPS) shares the clinical features of VWS with the addition of webbed skin of the legs, genital hypoplasia and/or oral synechiaes (MIM #119500).1 Both these autosomal dominant syndromes display a high degree of inter- and intra-familial phenotypic variation. The interferon regulatory factor 6 gene (IRF6) on 1q32.2 was identified by Kondo et al2 as the first disease gene responsible for VWS and PPS. A second VWS locus has been assigned to 1p34 by linkage in a large Finnish kindred.3 The IRF6 gene is 19 kb in size and includes 10 exons, out of which exons 3–9 are coding for a 467-amino-acid (aa) transcription factor. Until today, IRF6 mutations have been reported in a total of 79 kindreds (65 VWS and 14 PPS) in populations of various origin such as Japanese, Korean, Thai, European and Turkish.4, 5, 6, 7, 8, 9, 10, 11 The mutations spread over exons 2–9, with the majority affecting the helix–turn–helix DNA-binding domain (aa 13–113) or the protein-binding/SMIR domain (aa 226–394). To further characterize the mutation spectrum, we have investigated the occurrence of inherited and possibly de novo IRF6 mutations in VWS and PPS patients from 17 kindreds originating from Finland, Sweden, Norway, Thailand and Singapore.
Subjects and methods
Patients
Pedigrees and clinical details for the 16 VWS and one PPS kindred studied are shown in Figure 1. Individuals who had one or more of the following features were diagnosed as affected: lower LP, CP, CL, CLP or H.1 For six kindreds (VWS-8, -11, -14, -15, 16, -17), the clinical information and positive linkage to the VWS locus on 1q32 have been previously reported,12, 13 while the other 11 kindreds were newly recruited. All blood samples were collected with informed consent and the study has been approved by the local ethical committees both in Finland and Sweden.

Pedigrees of the 17 VWS and PPS families included in the present study. VWS-8 and -11 were first described as families 1 and 2 in Wong et al13 and VWS-14 to -17 as families 59–62 in Wong et al.12 Of all individuals whose symbol is filled black in the left bottom corner (cleft lip and palate/cleft lip), four present cleft lip (CL) only. These are in VWS-1, -6 and -11 as well as the second child in VWS-9. Individuals screened for mutation in the IRF6 gene using direct sequencing are indicated by ‘+’ and ‘−’ symbols. The presence of an IRF6 mutation is symbolized by a ‘+’ while ‘−’ denotes the absence of mutation. The nonsynonymous polymorphism p.Val274Ile (c.820G>A, rs17015215) in exon 7 was considered in all individuals screened for mutations. Its heterozygous form is indicated by ‘Val/Ile’ and its homozygous mutant form by ‘Ile/Ile’. All other screened individuals are wild-type (wt) homozygotes (Val/Val).
IRF6 mutation detection
In all, 32 affected and 26 clinically unaffected family members were screened for IRF6 mutation by direct sequencing of both DNA strands (Figure 1). Genomic DNA was extracted from peripheral leukocytes using a standard nonenzymatic extraction method based on salting out the cellular proteins by dehydration and precipitation with a saturated NaCl solution.14 Exons 1–9 were amplified by PCR as nine different fragments of 373–622 bp in size. The same primers were used for PCR and sequencing, and their sequences are available on request. Exons 1–8 were amplified using primers derived from within flanking intronic sequences, while for exon 9 the coding part and following 209 bp of the 3′ UTR were targeted. PCR was performed in 10 μl reactions containing 5 ng DNA, 3 mM MgCl2, 0.8 μ M of each oligonucleotide, 0.2 mM of each dNTP and 0.03 U/μl of polymerase. Exons 2–8 were amplified with Hot Star Taq™ polymerase (Qiagen), exon 1 with AmpliTaq DNA polymerase (Applied Biosystems) and exon 9 with Expand High Fidelity PCR system (Roche). Reactions were heated at 95°C for 15 min, subjected to 30–35 cycles of amplification (30 s at 95°C, 30 s at 50–63°C and 30 s at 72°C) and a final extension of 10 min at 72°C. Unincorporated oligonucleotides and dNTPs were removed by incubation with 0.4 U/μl of exonuclease I (New England Biolabs) and 0.072 U/μl shrimp alkaline phosphatase (Amersham Biosciences) during 30 min at 37°C. All exons were sequenced on both strands using the DYEnamic ET Dye terminator kit (Amersham Biosciences), following the manufacturer's instructions. Cleaned sequencing products were injected for 80 s at 3 kW and electrophoresed for 100 min at 9 kW or for 180 min at 6 kW using MegaBACE long read matrix on a MegaBACE 1000 instrument (Amersham Biosciences). Sequences were analyzed with the Sequence Analyser v3.0 software (Amersham Biosciences). To detect sequence variations, the chromatograms were inspected visually by two independent readers and sequences were aligned using the Pregap and Gap4 softwares from the Staden package (http://staden.sourceforge.net).
Genotyping of single nucleotide polymorphisms
The parents and the affected child in kindreds VWS-4 and -6 were genotyped for 20 single nucleotide polymorphisms (SNPs) using methods previously described.15, 16 All SNPs (rs6547203, rs12466369, rs1563314, rs11681515, rs10193323, rs7571685, rs10198233, rs7580922, rs4303736, rs1356218, rs12713962, rs4488689, rs10195905, rs1239063, rs7579479, rs1239070, rs1239080, rs1239064, rs1239085 and rs11126677) are located on 2p13.2 (www.snp.cshl.org) and were previously shown to be polymorphic in a Caucasian sample set of 200 individuals (H Anthoni, 2004, unpublished results).
Results
IRF6 mutations
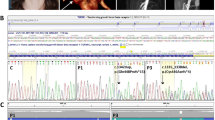
All nine exons and flanking intronic sequences of the IRF6 gene were fully sequenced in 58 individuals (32 affected and 26 unaffected) representing 16 VWS and one PPS kindred (Figure 1). The mutations identified in nine VWS and one PPS kindred are detailed in Table 1 and illustrated in Figure 2. All detected mutations are different, and to our knowledge, six of them are novel and have not been previously reported. The 10 identified mutations are located in exons 3, 4, 7, 8 or 9 of IRF6 and were all detected as heterozygous variations (Figure 2). The single base substitutions resulting in p.Lys34Glu and p.Arg84Cys affect the DNA-binding domain of the IRF6 protein, while the protein-binding domain is affected in p.Arg250Gln. The substitution p.Arg400Trp affects the carboxy-terminus of the protein. The other mutations include four nonsense mutations (p.Gln120X, p.Gln359X, p.Arg412X and p.Ser424X), and two deletions resulting in frameshifts (p.Gln118fs and p.Ser407fs). The p.Gln118fs that involves a 2 bp deletion in exon 4 (c.353_354delAG) is predicted to alter Gln118 and the following 17 aa, and then introduce a stop codon 53 bp downstream in exon 5. Similarly, p.Ser407fs is caused by a 1 bp deletion (c.1219delT) changing 29 aa from Ser407 in exon 9 and introducing a premature stop codon 89 bp downstream.

Sequencing chromatograms showing de novo (a) and inherited (b) IRF6 mutations detected in nine VWS and one PPS families. For each family the mutated sequence from an affected member (left) and the wt sequence of an unaffected member (right) are shown. The location of the mutation (vertical bar) and resulting protein change are indicated. Substitutions and deletions are indicated above the left chromatograms with the mutated base/sequence below the wt one. All chromatograms display the forward sequence on the coding DNA strand, except for VWS-8 where the reverse sequence from the noncoding strand is shown.
In six kindreds with detected mutation, more than one affected member was sequenced, which always showed the same mutation as in the proband. In two of these kindreds, PPS-2 and VWS-10, the mother and the maternal grand-mother of the proband, respectively, display the IRF6 mutation but only have H as clinical feature of the syndrome (Figure 1).
Other IRF6 polymorphisms detected
Two different polymorphisms were also identified and documented on both DNA strands. Both variants have been previously reported to be polymorphims by Kondo et al.2 The p.Val274Ile polymorphism (c.820G>A, rs17015215) in exon 7 was found in four unaffected members of three families (PPS-2, VWS-3 and -9) and in two affected individuals in VWS-9 and -3 (Figure 1). Kindreds VWS-3 and -9 are of Asian origin (Thailand and Singapore, respectively) where the frequency of the A allele is 22%, as compared to 3% in populations of European descent.
In exon 2, a common noncoding polymorphism, rs861019 (c.−73A>G), was also detected with allele frequencies of 60% (A) and 40% (G), similar to frequencies reported in the SNP database (www.snp.cshl.org). This variant does not seem to introduce any new splice site or start codon, and was not found to correlate with the VWS/PPS phenotype.
De novo IRF6 mutations
Three of the 10 mutations were found in kindreds where only the proband carried this variant (VWS-3, -4 and -6; Table 1). For two of these (VWS-4 and -6), samples from both parents were available for sequence and genotype analyses. In all, 12 and 19 of the 20 genotyped SNPs were informative in the respective kindreds. Haplotype reconstruction for each family was fully consistent with Mendelian inheritance, thus confirming the family relatedness. As the IRF6 mutation observed in the affected child was not present in any of the parents, they represent de novo mutations (Figure 2a and Table 1). It can be noted that both mutations (p.Gln120X and p.Gln359X) affect a glutamine residue and result in protein truncation following the introduction of a premature stop codon.
Lack of IRF6 mutation in the Finnish VWS kindreds
In all, 15 of the 17 kindreds in the study are of Nordic origin, including two Norwegian, nine Swedish and four Finnish. While mutations were detected in Norwegian (2/2) and Swedish (6/9) kindreds, no mutation was revealed in the Finnish ones (0/4) (Table 1).
Discussion
The present finding of 10 coding variants in VWS and PPS kindreds of Swedish, Norwegian, Thai and Singapore origin confirms the involvement of IRF6 in the pathogenesis of these allelic syndromes. The fact that six of those mutations are novel demonstrates that new mutations often occur in the IRF6 gene, for which 68 distinct mutations had already been detected in 126 VWS and 16 PPS kindreds screened.2, 4, 5, 6, 7, 8, 9, 10, 11 Taken together with the available literature, the present study expands the mutation spectrum to a total of 74 different IRF6 mutations identified in 89 kindreds (74 VWS and 15 PPS). The 89 mutations reported constitute nine deletions of 1–17, 162 bp, four insertions of 1–4 bp, two insertion/deletions and 74 base substitutions including 56 missense and 18 nonsense mutations.
The hitherto reported mutations are not randomly distributed in the IRF6 gene. In all, 42% (38/89) are located in the DNA-binding domain (DNA-BD) and 27% (24/89) are located in the protein-binding domain (protein-BD). The 300 bp ORF encoding the DNA-BD seems to be particularly prone to mutations. If mutations would be equally distributed over the 1401 bp IRF6 cDNA, 19 mutations would be expected as compared to the 38 observed. Similarly, in the 36 bp coding for the first 12 aa of the protein, seven mutations have been described while only two would be expected. The other coding parts of IRF6 are thus ‘undermutated’ (observed mutation number lower than the expected), with the least mutated part being the portion between the two conserved domains (nt 342–675; aa 114–225). While 21 mutations would be expected, only 10 were observed in the 89 kindreds with demonstrated IRF6 mutations.
The PPS kindred studied here carried a missense mutation c.250C>T. This mutation is the most common mutation and has been found in seven pedigrees,2, 5 including the present study. Six of these kindreds are phenotypically described as PPS and one as VWS. However, as both VWS and PPS display incomplete expressivity, one cannot exclude that the kindred described by Matsuzawa et al5 is not of PPS phenotype. As a matter of fact, the mother of our proband in the PPS-2 kindred was shown to carry the same mutation as her affected child and had only H as the sole sign of PPS phenotype. Further investigations would be necessary to evaluate the possibility of her being a mosaic carrier and therefore explaining her mild phenotype.
In two of the studied kindreds, the mutation was present in the proband but absent from both parents in spite of Mendelian inheritance of other DNA polymorphisms. Thus, these cases represent de novo mutations, a finding that has not been previously documented for IRF6 in VWS or PPS. No correlation between this type of event and some specific phenotypic trait or disease severity could be observed. The finding is well in agreement with the wide variety of IRF6 mutations found in mutation-carrying kindreds. Similarly, the recurrent observation of PPS and VWS patients with affected relatives in successive generations (but not in previous generations), has anticipated the existence of de novo mutation carriers.
No IRF6 variant was observed in any of the four Finnish VWS kindreds tested, as judged from sequencing of the CDS, the 5′UTR and the splice junctions. This finding was unexpected since three of them (VWS-14, -15 and -16) showed suggestive linkage to the 1q32–q41 region and all fullfill the criteria of VWS.12 The four pedigrees do have a strong genetic component, as three generations were documented with affected individuals in at least two generations. However, the fact that only one individual per kindred displays LP (together with CP or CLP) could be a sign that other affected, but distantly related, individuals diagnosed with only CP or CLP would in fact be phenocopies. Given the dominant pattern of inheritance observed in these pedigrees, this seems very unlikely. Moreover, no signs or symptoms of any other syndrome could be detected. Several explanations could clarify these findings. First, it is quite possible that a distinct IRF6 mutation is present as a founder mutation in all Finnish kindreds. This mutation could be located in the promoter region of the gene or in uncharacterized intronic regulatory sequences located outside the regions screened in our study. Large intragenic deletion in the IRF6 gene could also be the cause for the observed phenotype and might have gone undetected using our screening method, for example, direct sequencing. Deletion of the entire gene could be excluded in at least one of these Finnish kindreds (VWS-15) as one silent heterozygous polymorphism (rs2013162, exon 5) was detected in the screened affected individual. Alternatively, involvement of another locus than IRF6 in some kindreds could explain part of the negative findings. Linkage to the 1p34 locus was excluded in one of these kindreds, namely VWS-17.3 Finally, supporting our negative findings in the Finnish population, as compared to the other populations tested, is the fact that Finnish VWS kindreds do present phenotypic differences such as a higher frequency of CP than CL or CL/P.17
The present study reveals IRF6 gene mutation in 59% (10/17) of the kindreds, which is in accordance with the original investigation by Kondo et al,2 reporting mutations in 55% (59/107) of the screened kindreds.
References
Gorlin R, Cohen Jr M, Hennekam R : Syndromes of the head and neck; in: Gorlin RJ, Cohen MM, Hennekam RCM (eds). Oxford monographs on Medical Genetics, 4th edn. Oxford: Oxford University Press, 2001, pp 775–777, 905–907.
Kondo S, Schutte BC, Richardson RJ et al: Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat Genet 2002; 32: 285–289.
Koillinen H, Wong FK, Rautio J et al: Mapping of the second locus for the Van der Woude Syndrome to chromosome 1p34. Eur J Hum Genet 2001; 9: 747–752.
Kayano S, Kure S, Suzuki Y et al: Novel IRF6 mutations in Japanese patients with Van der Woude Syndrome: two missense mutations (R45Q and P396S) and a 17-kb deletion. J Hum Genet 2003; 48: 622–628.
Matsuzawa N, Yoshiura K, Machida J et al: Two missense mutations in the IRF6 gene in two Japanese families with Van der Woude syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2004; 98: 414–417.
Kim Y, Park JY, Lee TJ, Yoo HW : Identification of two novel mutations of IRF6 in Korean families affected with Van der Woude syndrome. Int J Mol Med 2003; 12: 465–468.
Shotelersuk V, Srichomthong C, Yoshiura K, Niikawa N : A novel mutation, 1234del(C), of the IRF6 in a Thai family with Van der Woude syndrome. Int J Mol Med 2003; 11: 505–507.
Wang X, Liu J, Zhang H et al.: Novel mutations in the IRF6 gene for Van der Woude syndrome. Hum Genet 2003; 113: 382–386.
Gatta V, Scarciolla O, Cupaioli M, Palka C, Chiesa PL, Stuppia L : A novel mutation of the IRF6 gene in an Italian family with Van der Woude syndrome. Mutat Res 2004; 547: 49–53.
Ghassibe M, Revencu N, Bayet B et al: Six families with van der Woude and/or popliteal pterygium syndrome: all with a mutation in the IRF6 gene. J Med Genet 2004; 41: e15.
Item CB, Turhani D, Thurnher D et al: Van Der Woude syndrome: variable penetrance of a novel mutation (p.Arg 84Gly) of the IRF6 gene in a Turkish family. Int J Mol Med 2005; 15: 247–251.
Wong FK, Koillinen H, Rautio J et al: Genetic heterogeneity and exclusion of a modifying locus at 17p11.2–p11.1 in Finnish families with van der Woude syndrome. J Med Genet 2001; 38: 198–202.
Wong FK, Karsten A, Larson O et al: Clinical and genetic studies of Van der Woude syndrome in Sweden. Acta Odontol Scand 1999; 57: 72–76.
Lahiri DK, Nurnberger JI : A rapid non-enzymatic method for the preparation of HMW DNA from blood for RFLP studies. Nucleic Acids Res 1991; 19: 5444.
Jurinke C, Van der Boom D, Cantor CR, Köster H : Automated genotyping using DNA MassARRAY technology. Methods Mol Biol 2002; 187: 179–192.
Peyrard-Janvid M, Anthoni H, Onkamo P et al: Fine mapping of the 2p11 dyslexia locus and exclusion of TACR1 as a candidate gene. Hum Genet 2004; 114: 510–516.
Lilius P : Epidemiology of cleft lip and palate in Finland 1975–1985. PhD thesis, University of Helsinki, 1992.
Acknowledgements
We thank Dr S Kondo for kindly providing us with some of the oligonucleotide sequences used in this study; and Dr Mimi Yow (National Dental Centre, Singapore) and Dr Pål Skaare (Rikshospitalet, Oslo, Norway) for collecting and providing us with the Singapore and Norwegian families, respectively. We are very grateful to the three anonymous reviewers for their comments, which have allowed us to improve the quality of our original manuscript. This work was supported by the Stockholm County Council, the Sigrid Jusélius Foundation, the Academy of Finland and the Swedish Research Council (‘Vetenskaprådet’).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Peyrard-Janvid, M., Pegelow, M., Koillinen, H. et al. Novel and de novo mutations of the IRF6 gene detected in patients with Van der Woude or popliteal pterygium syndrome. Eur J Hum Genet 13, 1261–1267 (2005). https://doi.org/10.1038/sj.ejhg.5201493
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201493
Keywords
This article is cited by
-
Tooth agenesis and orofacial clefting: genetic brothers in arms?
Human Genetics (2016)
-
Clinical and molecular findings in a Moroccan patient with popliteal pterygium syndrome: a case report
Journal of Medical Case Reports (2014)
