
Abstract
In thyroid eye disease, autoimmune inflammation of orbital musculature and fat increases the bulk of the orbital contents. Orbital tension rises and patients stratify according to the ease with which their globes can proptose. Restriction of proptosis is associated with optic nerve compression and visual loss; exophthalmos, with corneal damage. Ocular motility is affected, initially by muscle inflammation; late in the disease, by fibrosis.
Extraocular factors, including thyroid endocrine disturbance, antigen release, infections, malignancies, and smoking, may trigger and drive the orbital myopathy.
The management of thyroid eye disease by the identification and treatment of drives, followed by immunomodulatory therapy, is discussed.
Fourteen patients with compressive optic neuropathy were treated with immunomodulation using intravenous methylprednisolone, oral prednisolone, and cyclosporin A, and followed up for a minimum of three years. All recovered their pre-morbid visual acuities and visual fields in both eyes.
Severe disturbances of ocular motility also recovered in 30 patients, treated with the same regime. In one subject, ocular motility normalised with intravenous steroids and cyclosporin A, but no oral prednisolone. Morbidity from the treatment was low.
Immunomodulation is a rational and successful method for managing optic nerve compression and disordered motility in this condition.
Similar content being viewed by others

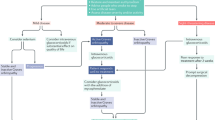

Natural history
In thyroid eye disease, an autoimmune response is followed by a cascade of pathology centred on orbital muscles and fat, but ultimately affecting oculomotor and visual function. Only medical management can be hoped to reverse an autoimmune disease; surgery, if required, reflects a failure of therapy and its role should be to ameliorate complications.
What is thyroid eye disease?
As thyroid eye disease begins, a lymphocytic infiltrate (consisting mainly of CD4+ lymphocytes) sweeps through the affected orbital muscles.1 When inflamed, they fail to contract and comply freely during eye movements, and pain and diplopia are early symptoms.
In this inflammatory phase of the disease, preadipocytes and fibroblasts, within the perimysium of rectus and oblique muscles and in orbital fat, secrete copious quantities of glycosaminoglycans.2 This hygroscopic material retains water, which causes the affected muscles to swell and further interferes with their function.
The orbit is a confined space and its bony surround is deficient only anteriorly. As the volume of its contents increases, orbital tension rises and patients stratify according to the ease with which this is relieved by proptosis:
Restricted proptosis/high orbital tension
(Figure 1)

(a) A 49-year-old type II diabetic patient with a toxic goitre, but adequate thyroid endocrine control, developed modest bilateral proptosis and lid retraction. After 5 months, her visual acuities fell to 6/36 (R), 6/9 (L) despite high doses of oral steroids and she underwent right surgical orbital decompression. After recovery from anaesthesia, she was found to have bilateral visual loss (CF both eyes; no colour awareness on R) and was transferred immediately to Addenbrookes Hospital. Both orbits were hard on retropulsion and a CT scan showed bilateral apical optic nerve compression. She received three pulses of intravenous methylprednisolone, oral prednisolone (30 mg/day), and cyclosporin A (plasma level of 200 μg/l). The intravenous methylprednisolone pulses were repeated after 1 month. After 10 months, her visual acuities had recovered to 6/12 (R), 6/9 (L) with full colour vision. (b) This visual function was retained at 5 years.
Proptosis may be prevented by unyielding lids and orbital septa, and by poorly compliant rectus muscles that anchor the globe to the orbital apex. Under these circumstances, the swelling rectus muscle bellies are forced into ever tighter proximity to the optic nerve, which they may compress directly where it enters the apex of the muscle cone. Orbital venous pressure can also rise, causing reduced pressure gradients across intraocular microcirculations, and disc oedema. Raised episcleral venous pressure is followed by elevation of intraocular pressure.
Proptosis/ocular surface damage
(Figure 2)

(a) Untreated thyroid eye disease: spontaneous decompression by proptosis, (b) attempted lid closure.
Alternatively, the lids and rectus muscles are sufficiently compliant to permit spontaneous anterior decompression of the orbit by proptosis. This may lead to lagophthalmos and corneal damage from exposure (particularly when non-compliant inferior rectus muscles splint the Bell's phenomenon).
In normal clinical practice, patients display a spectrum of responses between the extremes of optic nerve compression and severe proptosis. It is worth noting that those with least exophthalmos are often at the greatest risk of compressive optic neuropathy.
Late in the disease, fibroblasts secrete collagen into the new glycosaminoglycan matrix. This contracts, rendering muscle restriction permanent.
The eye is not the only organ to be affected by this process and excess mucopolysaccharide may also be laid down in the skin (pre-tibial myxoedema) and the nail beds (acropachy).
Why does thyroid eye disease arise?
Immunopathogenesis
Any theory to explain the pathogenesis of thyroid eye disease must consider why orbital myopathy occurs (briefly and reversibly) in the majority of patients with acute Graves' disease, but progresses in only a few. It must also resolve how thyroid eye disease can arise in patients whose endocrine disease is yet to come (or might never materialise), and may continue in those whose endocrine disease has remitted.
These disparate observations are best reconciled by an autoimmune disease, in which a single autoantigen is targeted in both the thyroid gland and the orbit. Current thinking favours the thyroid-stimulating hormone (TSH)-receptor, itself, as the shared antigen in most cases,1, 2, 3 but it is proposed that the response to autoimmunity is very different in the two sites.
When the autoantibodies that cause Graves' disease bind TSH-receptor in the thyroid gland, they directly stimulate thyroxine secretion. This bypasses the normal control of thyroxine production by TSH, released from the anterior pituitary gland under negative feedback inhibition from circulating thyroxine.4
TSH receptor is secreted, not only by cells of the thyroid gland, but also by orbital fibroblasts.2 When these are targeted by TSH-receptor antibodies, it is proposed that they respond by proliferation,5 glycosaminoglycan deposition, and fibrosis.
Fibrosis
The activation of fibroblasts to secrete glycosaminoglycans, then collagen, is controlled by cytokines released from tissue macrophages and migrating lymphocytes. Many have been implicated (including IGF-1, PDGF, TNFα, TGFβ), but TNFα and TGFβ appear to be particularly important.
Medical management
If this model for the aetiology of thyroid eye disease is sound, medical management must abort an immune response that is already vigorous when a patient presents. The removal of antigen is rarely an option in autoimmune disease, but it may be possible to limit its availability. Other nonspecific drives (particularly, smoking, bacteria, and tumours) can also be identified and suppressed. Only then does immunomodulation become appropriate.
The regime presented here aims briefly to arrest the cycle of autoimmunity, then subdue its recrudescence. The need to inhibit fibroblast activation and collagen secretion is also addressed.
Identification and control of triggers and drives
Endocrine
Both hyper- and hypo-thyroidism can cause thyroid orbital myopathy to progress. In the latter case, increased secretion of TSH-receptor (autoantigen) by thyroid follicular cells6 may be responsible. Therefore, careful management of thyroid endocrine disease is essential before attempting to treat the orbital myopathy by immunomodulation.
The adequacy of endocrine control is estimated from the plasma TSH level, which alters slowly, but best reflects the availability of the active hormone, tri-iodothyronine, in tissues. A block–replace regime (in which thyroxine production is completely aborted by excess carbimazole or propylthiouracil, and administered L-thyroxine is titrated against the plasma TSH level) gives more stable control of thyroid function than does the simple titration of carbimazole dose against plasma TSH. In the latter regime, it is likely that short-term fluctuations in endogenous thyroxine secretion arise, but fail to be reflected by TSH on account of the longer period of its response.
It is conventional endocrine practice to withdraw a block–replace regime after 6 months in patients with a first episode of thyrotoxicosis.7 About 60% require no further treatment and those in whom disease recurs usually receive radioactive iodine (131I) or thyroidectomy. In patients with thyroid eye disease, a recurrence of thyrotoxicosis risks reactivating the orbital myopathy; therefore, the block–replace treatment should continue until thyroid eye disease has been inactive clinically for at least 6 months.
Antigen release from the thyroid
Thyroid destruction by 131I may trigger or exacerbate thyroid eye disease,8 presumably as a consequence of massive antigen release. It is a matter of anecdote that surgical thyroidectomy can occasionally have a similar effect. Either procedure should be avoided in patients with active orbital myopathy and covered by systemic steroids in those whose orbital disease is inactive.
Infection and malignancy
Remote infection or malignancy can enhance autoimmunity and the medical management of thyroid eye disease should begin by identifying and eradicating these triggers and drives. There are probably many mechanisms, but superantigenicity is the best understood. Certain molecules (prominently, bacterial endotoxins produced by Staphylococcus aureus) interact with MHC molecules causing nonspecific facilitation of antigen recognition and activation of memory T-lymphocytes.9
Smoking
Smoking of tobacco enhances a number of autoimmune diseases and has been shown to increase the risk that patients suffering from hyperthyroidism develop thyroid eye disease.2 The author has encountered many patients in whom active thyroid eye disease remitted following the cessation of smoking and no other intervention.
Interruption of the autoimmune cycle
An organ-specific autoimmune disease may be considered to be a cycle of: autoantigen presentation and recognition, expansion of autoreactive lymphocyte clones, antibody-mediated or T-cell mediated cytotoxicity (with damage to the target organ), and release of more autoantigen.
By the time a patient presents with thyroid eye disease, this cycle is in full swing and has to be interrupted. A simple expedient is the administration of high-dose intravenous steroids, which halt lymphocyte recirculation and interfere with inflammation and the release of autoantigen.
Maintenance of remission
Cyclosporin A and tacrolimus inhibit calcineurin, preventing the secretion of interleukin-2 by CD4+ T-lymphocytes,10 and thereby interfering with the expansion of lymphocyte clones. Acting on the amplification phase of the immune response, they are well placed to subdue recrudescence of the cycle of autoimmunity.
Inhibition of fibrosis
A number of cytokines; particularly, TNFα which is secreted by roaming CD4+ lymphocytes and tissue macrophages, drive fibroblasts to proliferate and secrete glycosaminoglycans, then collagen. Conveniently, TNFα release from either source is inhibited by cyclosporin A and tacrolimus,11 making these also ideal agents to prevent the cicatricial phase of thyroid eye disease.
Control of thyroid eye disease with intravenous methylprednisolone and cyclosporin A
Compressive optic neuropathy
Subjects
Fourteen patients (12 female, two male), aged between 33 and 78 years at presentation (mean 54 years) participated in the study. They attended Addenbrooke's Hospital between 1990 and 2000 and were followed up for at least 3 years. All had evidence of optic nerve compression by at least one of the criteria given in Table 1.
Methods
Baseline observations were performed (Tables 2 and 3) to assess disease activity and identify drives. Where possible, treatment of these drives was initiated before the introduction of immunomodulation (Table 4). Every patient then underwent 3 months of frequent clinical observations.
Full clinical assessments (Tables 2 and 3) took place at intervals of 4 months. At 0, 7, 30, 90, 180, 360, 720 and 1080 days, after the initiation of treatment, data for visual acuity and visual fields (mean deviation) were recorded on spread-sheets.
In 11 subjects who had been treated for optic nerve compression, data for field of binocular single vision (BSV) were also available. This indicated substantial resolution of diplopia. The same regime was therefore used on a further 19 patients, all of whom had severe diplopia, but no optic nerve compression. Fields of BSV were quantified12 at 0, 180, 360, and 720 days after starting treatment and at the most recent clinical assessment (between 3 and 6 years).
Results
Visual acuity Acuity was restored to normal unless there had been pre-existing visual impairment (Figure 3a and b).

Visual acuity (Snellen): (a) right eyes, (b) left eyes. Fourteen patients with compressive optic neuropathy: 0, 7, 30, 90, 180, 360, 720, and 1080 days after intravenous methylprednisolone and the initiation of oral prednisolone and cyclosporin A.
Visual fields Mean deviations of automated visual fields recovered in every patient (Figure 4a and b).

Visual fields (Humphrey: mean deviation): (a) right eyes, (b) left eyes. Fourteen patients with compressive optic neuropathy: 0, 7, 30, 90, 180, 360, 720, and 1080 days after intravenous methylprednisolone and the initiation of oral prednisolone and cyclosporin A.
Motility Clinically significant improvements occurred in every patient except one. Recovery was slower than for visual acuity and fields (sometimes as late as 2 years). By 3 years, all patients were asymptomatic or controllable with prisms and only one required strabismus surgery (Figure 5).

Fields of binocular single vision (BSV). Thirty patients with diplopia: scores from 0, 180, 360, and 720 days after treatment, and the most recent clinic assessment (between 3 and 6 years). All patients were treated with intravenous methylprednisolone, oral prednisolone, and cyclosporin A.
Side effects of treatment See Table 5.
Discussion
Thyroid eye disease is an autoimmune orbital myopathy and it is not surprising that it responds well to immunomodulatory therapy. As with other autoimmune diseases, successful management requires the physician to identify and avert or treat triggers and drives, which here include endocrine disturbance, antigen availability, smoking, infections, and malignancy.
The patients who were recruited into the first phase of this study had severe compressive optic neuropathy, which would have been managed by surgical decompression in most other centres.
In the regime that is presented here, intravenous steroids are used not only to arrest the vigorous autoimmune response that is under way when thyroid eye disease presents, but also to reduce orbital oedema. The rapid recovery of visual acuity and field loss is testament to their effectiveness. Cyclosporin A is chosen both to control autoimmunity and to inhibit late fibrosis, and the recovery of ocular motility in a cohort of 30 patients confirms that this, too, is effective.
Medical regimes to control thyroid eye disease conventionally use high doses of oral steroids. The dose used in this study was modest, but some side effects did still arise, of which hypertension was the most common; gastrointestinal haemorrhage and myocardial infarction, the most severe. However, it must be remembered that this group of patients was followed for 3–6 years and most had a long history of cigarette smoking, which may equally have been responsible.
One patient with severely disturbed ocular motility, having accepted intravenous methylprednisolone, refused all oral steroids and elected to rely on cyclosporin A alone. Her field of BSV still recovered fully and it is proposed that oral steroids may be reduced further, or even omitted, in future treatment regimens that combine intravenous steroids with cyclosporin A.
References
Heufelder AE . Pathogenesis of ophthalmopathy in autoimmune thyroid disease. Rev Endocr Metab Disord 2000; 1 (1–2): 87–95.
Ludgate M, Baker G . Unlocking the immunological mechanisms of orbital inflammation in thyroid eye disease. Clin Exp Immunol 2002; 127 (2): 193–198.
Ludgate M, Crisp M, Lane C, Costagliola S, Vassart G, Weetman A et al. The thyrotropin receptor in thyroid eye disease. Thyroid 1998; 8 (5): 411–413.
Weetman AP . Autoimmune Endocrine Disease. Cambridge University Press: Cambridge, 1991.
Feldon SE, Park DJ, O'Loughlin CW, Nguyen VT, Landskroner-Eiger S, Chang D et al. Autologous T-lymphocytes stimulate proliferation of orbital fibroblasts derived from patients with Graves' ophthalmopathy. Invest Ophthalmol Vis Sci 2005; 46 (1): 3913–3921.
Huber GK, Concepcion ES, Graves PN, Davies TF . Positive regulation of human thyrotropin receptor mRNA by thyrotropin. J Clin Endocrinol Metab 1991; 72 (6): 1394–1396.
Weetman AP, Pickerill AP, Watson P, Chatterjee VK, Edwards OM . Treatment of Graves' disease with the block-replace regimen of antithyroid drugs: the effect of treatment duration and immunogenetic susceptibility on relapse. Q J Med 1994; 87: 337–341.
Bartalena L, Tanda ML, Piantanida E, Lai A, Pinchera A . Relationship between management of hyperthyroidism and course of the ophthalmopathy. J Endocrinol Invest 2004; 27 (3): 288–294.
Chatila TA, Geha RS . Superantigens. In: Lachmann PPJ, Peters DK, Rosen FS and Walport MJ (eds). Clinical Aspects of Immunology, 5th ed. Blackwell: Boston, 1993.
Ho S, Clipstone N, Timmermann L, Northrop J, Graef I, Fiorentino D et al. The mechanism of action of cyclosporin A and FK506. Clin Immunol Immunopathol 1996; 80: S40–S45 (Review).
Losa Garcia JE, Mateos Rodriguez F, Jimenez Lopez A, Garcia Salgado MJ, Martin de Cabo MR, Perez Losada J et al. Effect of cyclosporin A on inflammatory cytokine production by human alveolar macrophages. Respir Med 1998; 92 (5): 722–728.
Fitzsimons R, Fraco, White FJ . Functional scoring of the field of binocular single vision. Ophthalmology 1990; 97: 33–35.
Acknowledgements
I thank John Ferris, Rachna Murthy, and Sarah Meredith, without whose energy and enthusiasm this study would never have been completed.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Meyer, P. Avoiding surgery for thyroid eye disease. Eye 20, 1171–1177 (2006). https://doi.org/10.1038/sj.eye.6702393
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.eye.6702393
Keywords
This article is cited by
-
Life-threatening complications of high doses of intravenous methylprednisolone for treatment of Graves’ orbitopathy
Thyroid Research (2019)
-
Immune reconstitution after alemtuzumab therapy for multiple sclerosis triggering Graves’ orbitopathy: a case series
Eye (2019)
-
Clinical pathophysiology of thyroid eye disease: The Cone Model
Eye (2019)
-
Interruption of autoimmunity for thyroid eye disease: B-cell and T-cell strategy
Eye (2019)
-
Serial TSH-receptor antibody levels to guide the management of thyroid eye disease: the impact of smoking, immunosuppression, radio-iodine, and thyroidectomy
Eye (2019)
