
Abstract
Allergic rhinitis (AR) is a complex disorder with a polygenic, multifactorial aetiology. Twin studies have found the genetic contribution to be substantial. We collected and clinically characterised a sample consisting of 127 Danish nuclear families with at least two siblings suffering from AR or allergic conjunctivitis including 540 individuals (286 children and 254 parents). A whole-genome linkage scan, using 424 microsatellite markers, was performed on both this sample and an earlier collected sample consisting of 130 families with atopic dermatitis and other atopic disorders. A third sib-pair family sample, which was previously collected and genotyped, was added to the analysis increasing the total sample size to 357 families consisting of 1508 individuals. In total, 190 families with AR was included. The linkage analysis software Genehunter NPL, Genehunter MOD, and Genehunter Imprinting were used to obtain nonparametric and parametric linkage results. Family-based association analysis of positional candidate SNPs was carried out using the FBAT program. We obtained genome-wide significant linkage to a novel AR locus at 1p13 and suggestive linkage to two novel regions at 1q31-q32 and 20p12, respectively. Family-based association analysis of SNPs in the candidate locus DNND1B/CRB1 at 1q31 showed no significant association and could not explain the linkage signal observed. Suggestive evidence of linkage was also obtained at three AR loci previously reported (2q14-q23, 2q23, and 12p13) and indication of linkage was observed at a number of additional loci. Likely maternal imprinting was observed at 2q23, and possible maternal imprinting at 3q28.
Similar content being viewed by others


Introduction
Allergic rhinitis (AR) is a growing problem worldwide. Today, 10–30% of adults and up to 40% of children suffer from this condition.1 However, the rise in prevalence appears to have reached a plateau in many western countries since the 1990s.2, 3, 4 The typical symptoms are drip, block, and itch of the nose along with sneezing due to inflammation of the nasal mucosa often accompanied by allergic conjunctivitis (itch and redness of the eye).1 AR is often mistakenly considered a disease of little importance. It is however a frequent cause of morbidity, medical treatment costs, lost work days, and reduced school performance.1 Allergic diseases are complex multi-factorial and polygenic disorders. The genetic contribution to AR has been shown to be substantial. Twin studies have shown heritability estimates of 71–96%.5, 6, 7 Considerable research has focused on the genetic basis of asthma and to some extent atopic dermatitis (AD), whereas AR has been somewhat neglected. Only four whole-genome scans and three regional linkage scans of the phenotype AR have been reported.8, 9, 10, 11, 12, 13, 14 More research has been performed in the field of candidate gene association analysis and more than 30 genes showing evidence of association with AR have been reported (Supplementary Table 1). However, the associations have rarely been unambiguously replicated in a number of populations. Currently, no genome-wide association studies (GWAS) specifically addressing AR in Caucasians have been published (http://www.genome.gov/GWAStudies/#1). In this study, we performed a whole-genome linkage scan of two samples: one sample containing 127 Danish nuclear families, each with at least two siblings suffering from AR or allergic conjunctivitis, and a second sample consisting of 130 Danish nuclear families with AD and other atopic disorders.15 The two samples were combined with a third sib-pair atopy family sample previously collected and genotyped,16 reaching a total of 357 families consisting of 1508 individuals of whom 190 families had two siblings with AR. We also performed a family-based association analysis of candidate DNND1B/CRB1 SNPs at 1q31.
Materials and methods
Subjects
Three family samples (A, B, and C) were included in the study. Sample A consisted of 127 Danish nuclear families with a total of 286 children and 254 parents. The recruitment of these families was performed through the paediatric departments in Viborg, Aarhus, Hjoerring, Herning, and Aalborg, as well as the Department of Respiratory Diseases, Aarhus University Hospital and one private paediatric clinic in Aalborg. All individuals were clinically examined and tested through a questionnaire by the same doctor. The questionnaire contained questions about symptoms, duration, and treatment of rhino-conjunctivitis, AD, and asthma. Atopic disease was diagnosed according to standard criteria.1 The diagnoses were evaluated by a second doctor who assessed the questionnaires without seeing the individuals.
The families were grouped according to phenotypes being present in at least two and as many as four of the siblings (Table 1). The mean age among the children was 16 years, and 52% were male. Blood was collected from all participants for DNA analysis and for serum measurements of total IgE and allergen-specific IgE antibody (ImmunoCAP system, Phadia Aps, Allerød, Denmark).
Samples B and C comprised two earlier collected Danish sibling-pair family samples. Sample B contained 130 Danish nuclear families with at least two full siblings with AD.15 The mean age among the children was 12.5 years, and 48% were male. Sample C contained 100 Danish nuclear families enroled in the Danish Allergy Project. Inclusion criteria were at least two full siblings with a doctor diagnosed atopy, such as asthma, rhinitis or AD.16 The mean age among the children was 14.5 years, and 49% were male. Thus, a total of 425 children with AR and/ or allergic conjunctivitis and 394 parents from 197 Danish nuclear families were analysed in this study (Table 1). Sample A is a novel sample, and sample B has not been reported previously in respect to AR. However, sample C has been reported previously.8 The parents in all three samples were also phenotypically assessed, and their affection status was included in the analysis. The Danish scientific ethical system approved the protocol. All participants and/or their parents signed informed consent forms.
Phenotype definition
The AR phenotype was studied using three approaches. The first two are broad definitions based purely on a clinical symptom-based diagnosis. The first of these approaches is named ‘Hay fever combined’ (HC), and this group contains subjects who present clinical symptoms of AR and/or allergic conjunctivitis; the second broad group contains subjects who have clinical symptoms of AR. The third group contains a more defined phenotype (IgE-associated AR), where the subjects have AR in combination with a positive specific IgE (defined as a positive specific IgE antibody for at least one of the tested allergens). The defined phenotype of this third group may decrease the genetic heterogeneity and thereby increase the statistical power of the study. The allergen-specific serum IgEs were tested for reactivity with 12 allergens (Supplementary Materials for details on allergens). A specific IgE ≥0.35 kU/l was considered elevated.
Genotyping
Genotyping was performed on genomic DNA from both children as well as their parents. Samples A and B were genotyped at the University of Antwerp, Belgium, using a panel of 424 microsatellite markers (Supplementary Table 2). The details from the quality control filtering can be found in Supplementary Materials. Sample B had previously been genotyped with 91 fine mapping markers on chromosome 3, 4, and 18, 16 of these were also within the Belgian panel (Supplementary Table 2); thus, 75 additional markers were included to increase statistical power of sample B. The degree of LD between markers on chromosome 3, 4, and 18 in sample B was assessed using the software GOLD (http://www.sph.umich.edu/csg/abecasis/GOLD/). Sample C was previously genotyped using 446 microsatellite markers.8 Sex-average Marshfield linkage maps were used for all analyses. For the association study, genotyping of candidate SNPs (rs2111931 in the DNND1B gene, and rs2786098, rs12134409, rs2786101, rs2821125, rs1337168, rs1337167 in the CRB1gene) was performed using the MassARRAY Genotyping system (Sequenom, San Diego, CA, USA) as described in Nyegaard et al.17 The DNND1B/CRB1 locus is characterised by high LD and all seven candidate SNPs showed strong pairwise LD (r2 from 0.76 to 1.00, CEU population), thus serving as surrogate markers for each other and the majority of common variation across the entire locus. Four SNPs failed genotyping due to failed PCR amplification in all samples. The remaining three SNPs had a call rate above 0.97. No deviation from Hardy–Weinberg equilibrium was seen among the parents for any of the SNPs. Power calculations performed in PBAT18 using the allelic odds ratios reported by Sleiman et al19 (0.63), and assuming a disease frequency of 0.1, showed that we should have a power of 1 to detect associations with all three genotyped SNPs in the combined A+B sample.
Statistical analyses
Linkage analyses were performed by the affected sib-pair method.20 Multipoint nonparametric linkage (NPL) analyses were performed using Genehunter NPL v2.0 for the autosomal chromosomes and Genehunter-Imprinting v1.321 for the X chromosome. A scan step size of five was applied. The allele frequencies were calculated from the parents. For chromosomes with NPL score ≥1.5, a parametric analysis was performed using Genehunter MOD and Genehunter-Imprinting.22 Because Genehunter MOD maximises the LOD score with respect to different disease-model parameters, the MOD scores are somewhat inflated and are not directly comparable to LOD scores.
Using Genehunter Imprinting, the ‘parent-of-origin effect’ was analysed with different penetrances for heterozygous individuals, depending on whether the disease-causing allele was transmitted by the father or the mother. Evidence of imprinting is found when (1) the difference between P(wt/d) and P(d/wt) exceeded 0.2,22 (2) the difference between imprinting and non-imprinting MOD scores exceed 1.5,23 and (3) the imprinting MOD was high enough to at least indicate evidence of linkage in the region.
To evaluate the genome-wide level of significance in our samples, empiric P-values were calculated for the parametric MOD scores using the simulation features incorporated in Genehunter MOD and Genehunter-Imprinting.21, 22 A total of 10 000 simulations were performed for each chromosome/region. For MOD scores equal to and above four, the number of simulations was increased to 100 000. Based on the regional empiric P-value (Pregional) and the size of the region tested, a genome-wide empiric P-value (Pgenome) for each MOD score was calculated as empiric Pgenome=empiric Pregion × (size of whole genome/size of simulated region). This Pgenome indicate the likelihood of obtaining a MOD score as high as or higher than the observed MOD score in a whole genome by chance. We used a threshold for suggestive linkage of P=0.0007 (corresponding to the observed result occurring by chance less than once in a whole genome scan) and for significant linkage P=0.00002 (occurring by chance less than 0.05 times in a whole genome scan).24 No correction for testing multiple phenotypes was performed as the three AR phenotypes tested are highly correlated. MOD score calculations on the X chromosome was not possible using Genehunter MOD or Genehunter Imprinting. Association analysis of the two family samples was carried out using FBAT version 2.03.25, 26 Because of the detected linkage signal in the area, we used the empirical variance option in the FBAT program, eliminating a possible linkage-driven inflation of association results caused by transmission of identical alleles in siblings.
Results
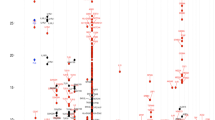
The results of the parametric and nonparametric analyses are summarised in Table 2. The NPL scores of chromosomes with NPL scores ≥2 in at least one of the three rhinitis phenotypes are illustrated in Figures 1 and 2.

NPL scores for sample A for all chromosomes where NPL>2 for one of the three phenotypes investigated.

NPL scores for the combined sample (sample A, B and C) for all chromosomes where NPL>2 for one of the phenotypes investigated. When all the three samples were combined, the results for the phenotypes HC and AR were almost identical.
In the parametric analysis, a region on 1p13 reached genome-wide significance (MOD 4.20; Pgenome=0.037) for HC in the combined sample. In addition, a number loci reached suggestive evidence of linkage to HC including 1q31 (imprinting MOD 3.38; Pregion=0.007), 2q23 (imprinting MOD 3.00; Pregion=0.076), 20p12 (MOD without/with imprinting 3.52/3.81; Pregion=0.0025/0.0033).
For the AR phenotype, suggestive evidence of linkage was found for the chromosomes 1p13 (MOD 2.97; Pregion=0.0055), 1q31 (imprinting MOD 3.10; Pregion=0.054), 2q14–q21 (imprinting MOD 3.33; Pregion=0.0045), 2q23 (imprinting MOD 3.26; Pregion=0.0077), 12p13 (imprinting MOD 3.10; Pregion=0.038), and 20p12 (MOD without/with imprinting 3.66/4.25; Pregion=0.0039/0.0036).
The phenotype IgE-AR revealed suggestive evidence of linkage at chromosome 1p13 (MOD without/with imprinting 4.01/4.01; Pregion=0.0006/0.0008), 1q32 (MOD without/with imprinting 3.16/3.28; Pregion=0.003/0.007), and 20p12 (imprinting MOD 3.30; Pregion=0.018).
In the nonparametric analysis, only chromosome 20p12 reached the level of suggestive linkage as defined by Lander et al.24 This was obtained for the combined sample for the phenotypes HC and AR (NPL 2.76; P=3.8 × 10−4 and NPL 2.76; P=3.7 × 10−4, respectively). Several loci reached or exceeded NPL 2.00 with nominal P-values less than 0.01, thereby showing ‘interesting’ evidence of linkage according to Haines et al27 (Table 2). These loci were chromosomes 1q21, 4q11-q12, 14q12, 20p13, 22q12-q13, Xp22, Xp21, and Xp11.
Table 3 summarises the MOD scores and penetrances for loci showing signs of imprinting. Most of the loci showed a difference less than the recommended 1.5 between the imprinting and the non-imprinting MOD scores. Furthermore, the difference between P(wt/d) and P(d/wt) were below 0.2. However, at locus 2q23 in the AR phenotype, a difference of 1.52 between the imprinting and the non-imprinting score and a P(wt/d)>P(d/wt) of 0.205 was found. At locus 3q28 in the IgE-AR phenotype, a fairly large difference between P(wt/d) and >P(d/wt) of 0.69 was found along with an imprinting MOD of 2.84 and a difference between the imprinting and non-imprinting MOD of 1.32. No clusters of high LD among the densely spaced fine mapping markers were found. Thus, the linkage signals identified were not inflated by high LD between the markers.
SNPs located in the DNND1B/CRB1 locus at 1q31 have previously been associated with asthma in a GWAS of two samples of European ancestry and one of African ancestry.19 To see whether the obtained linkage signal at 1q31 could be explained by these polymorphisms, we conducted a family-based association analysis in the combined sample (A+B) with the three most significantly associated, genotyped SNPs from the GWAS19 (rs2786098, rs12134409, and rs2111931) as well as the four most significantly associated, imputed SNPs (rs2821125, rs1337168, rs1337167, and rs2786101). The seven SNPs are all in relatively tight intermarker LD in the European populations, where the association was identified.19 The three SNPs passing quality control (rs2786098, rs12134409, rs2786101) did not show any significant associations in our samples. The SNP that was most strongly associated with asthma in the GWAS (rs2786098, major allele more frequent among cases) showed a weak trend towards over-transmission of the major allele in IgE-associated AR (P=0.18) (Supplementary Table 3). Thus, the candidate DNND1B/CRB1 SNPs could not explain the linkage signal and do not seem to have a major role in AR susceptibility.
Discussion
The present genome scanning of three Danish family samples revealed a number of loci showing evidence of linkage to AR. Chromosome 1p13 reached genome-wide significance, whereas suggestive evidence of linkage was found on chromosome 1q, 2q, 12p, and 20p. NPL above 2 was obtained in regions on chromosome 1q, 4q, 14q, 20p, 22q, and Xp. Highest MOD scores were found at 1p13 and 20p12.
Chromosome 1p13 has not been reported earlier in connection with AR or atopy. The locus has previously shown suggestive linkage to asthma.28 Possible candidate genes are the glutathione S-transferase M1 (GSTM1)29 and acidic mammalian chitinase (CHIA) genes located at 1p12-p13, earlier associated with asthma.30 Chromosome 20p12 is also a novel finding in regard to AR, however, it is earlier found to be linked to atopy and asthma.16, 28 No obvious candidate gene is found in the region.
Three of the regions with suggestive linkage to AR in our study have been reported previously (2q14-q21, 2q23, and 12p13) as well as two of the regions with weaker linkage to AR (22q12-q13 and Xp21).8 These regions were identified from a linkage scan performed by Haagerup et al.8 The study sample from Haagerup et al8 was included in our analysis as sample C. Only linkage obtained at 2q23 was based solely on sample A, the remaining loci were found when analysing the combined phenotype. Thus, these regions do not represent an independent linkage replication, although the evidence for linkage has increased by increasing the sample size. Region Xp11 and 14q12 have been reported earlier in association with allergic sensitisation and asthma, respectively.16, 28, 31
AR is most often studied as a subgroup of another allergic disease and seldom is AR the main focus of the study. We focused on AR when collecting our sample A, increasing the power to detect primarily AR-related loci. The participants in our study also suffered from other atopic diseases. However, they did not all have the same atopic manifestations along with AR. Thus, whereas the present study power is much higher to identify AR loci rather than loci specific for other (comorbid) allergic phenotypes, some of the loci reported may well be shared with other allergic phenotypes.
Only four whole-genome scans of the phenotype AR8, 9, 10, 11 and three regional linkage analyses of parts of the genome12, 13, 14 have been reported (Supplementary Table 4). For all published AR genome-wide scans (except the study by Yokouchi et al9), the samples were originally collected focusing on other atopic diseases than AR. The AR phenotype was then analysed as a phenotypic subgroup thereby potentially increasing the selection bias and genetic heterogeneity between the studies. Our study is one of the first to focus primarily on the AR phenotype. When comparing AR-linked regions in our sample to published independent studies, only sparse overlap in the AR regions are found, which may partly be explained by the difference in study design and phenotype assessment. The phenotype in one study was based solely on answers from a standardised questionnaire,10 whereas other studies have based the phenotype on doctor-confirmed diagnoses.8, 9, 11 Furthermore, one study examined the phenotype AR,10 whereas the three other genome-wide studies investigated IgE-AR.8, 9, 11 Additionally, power limitations and variations in statistical methods may cause discrepancies in the reported findings.
Our results showed an absolute absence of a signal in the region of 3q13 for AR. This region showed a highly significant linkage in a fine scale linkage study by Brasch-Andersen et al14 who also included the sample we have included as sample C. A possible explanation may be that the subjects in this study almost all had asthma as well as AR. In our study, only 29% and 42% of the AR sib-pairs in samples A and B, respectively, had co-existing asthma. Thus, the signal previously reported at chromosome 3q13 may primarily be linked to AR in the context of asthma, rather than to AR alone.
More than 30 genes showing association with AR has been reported (Supplementary Table 1). Four of these candidate genes reside in regions highlighted in this study, including the filaggrin gene (FLG) gene on 1q21. Several studies have found an association of FLG with AD and atopy as well as AR and asthma when co-excising with AD.32 The FLG gene is positioned 7 Mb from the maximum linkage signal, a span containing almost a hundred genes, some of which have earlier been linked to atopic conditions.33, 34 At 2q13-q14, the interleukin 1 gene cluster (IL1A, IL1B and IL1RN) previously associated with AR resides.35 Although the cluster is positioned as far as 16 Mb from the maximum linkage signal obtained, this span only contains approximately fifty confirmed genes. At 20p13, a disintegrin and metalloproteinase domain 33 (ADAM33) gene residing almost 3 Mb from the maximum linkage signal has previously been associated with AR.36 Toll-like receptor 7 and 8 (TLR7 and TLR8) located at Xp22 confer susceptibility to several allergic diseases and among these are AR.37 TLR7 and TLR8 are positioned just over 8 Mb from the maximum linkage signal, a span containing only about 25 confirmed genes. Beside genes associated to AR, several of the highlighted regions contains genes previously associated with asthma19, 29, 38, 39, 40 or atopy.41, 42, 43
Inactivation of either the paternal or maternal copy of a gene is known as imprinting. Imprinting may be due to either DNA methylation or changes in the chromatin structure. The obtained imprinting MOD score of 3.26 at locus 2q23.3 supports the presence of imprinting. At locus 3q28 in the IgE-AR phenotype, only a moderate difference of 1.32 was found between the MOD score with and without imprinting. The MOD score obtained with imprinting is high, suggesting a possible maternal imprinting may exist (Table 3). We are not aware of any previous allergy-related linkage studies reporting imprinting in these regions.
This study has limitations. It must be noted that we used sex-average maps for all analyses, which may influence the result of the imprinting analysis. The use of sex-specific maps would have been more ideal, as imprinting MOD scores can be biased when a sex-averaged instead of the correct sex-specific map is employed, however simulation studies show that this bias is limited when both parents are genotyped,44 as in our study. Also, although the three rhinitis (sub) phenotypes analysed are highly correlated, there is a minor degree of multiple phenotype testing inflating the significance of the results. The precise extent of inflation however cannot be estimated without extensive simulations.
In conclusion, our genome-wide linkage scan using affected sib-pairs has identified several regions that may contain a gene involved in the pathogenesis of AR. Significant linkage was obtained at a novel AR locus at 1p13. Candidate SNPs in the DNND1B/CRB1 locus at 1q31 showed no significant association and could not explain the linkage signal observed. Among the regions suggestive of linkage, two were novel regions (1q31-q32 and 32p12) and three were previously reported (2q14-q23, 2q23, and 12p13) with only 2q23 representing an independent replication because of sample overlap for the remaining findings.8 Our findings may direct the search for susceptibility genes by providing a basis, for example, association analysis of positional candidate genes, regional fine mapping using tagSNPs, prioritisation of regional SNPs in GWA studies, and targeted next-generation sequencing approaches. Identification of a risk gene for AR or other atopic diseases may increase our knowledge of the pathophysiology of these complex diseases and pave the way for more precise disease-classification and diagnosis, as well as facilitate more personalised treatments.
References
Wallace DV, Dykewicz MS, Bernstein DI et al: The diagnosis and management of rhinitis: an updated practice parameter. J Allergy Clin Immunol 2008; 122: S1–84.
Zollner IK, Weiland SK, Piechotowski I et al: No increase in the prevalence of asthma, allergies, and atopic sensitisation among children in Germany: 1992-2001. Thorax 2005; 60: 545–548.
Braun-Fahrlander C, Gassner M, Grize L et al: No further increase in asthma, hay fever and atopic sensitisation in adolescents living in Switzerland. Eur Respir J 2004; 23: 407–413.
Gupta R, Sheikh A, Strachan DP, Anderson HR : Time trends in allergic disorders in the UK. Thorax 2007; 62: 91–96.
Thomsen SF, Ulrik CS, Kyvik KO, Skadhauge LR, Steffensen I, Backer V : Findings on the atopic triad from a Danish twin registry. Int J Tuberc Lung Dis 2006; 10: 1268–1272.
Rasanen M, Laitinen T, Kaprio J, Koskenvuo M, Laitinen LA : Hay fever - a Finnish nationwide study of adolescent twins and their parents. Allergy 1998; 53: 885–890.
van Beijsterveldt CE, Boomsma DI : Genetics of parentally reported asthma, eczema and rhinitis in 5-yr-old twins. Eur Respir J 2007; 29: 516–521.
Haagerup A, Bjerke T, Schoitz PO, Binderup HG, Dahl R, Kruse TA : Allergic rhinitis - a total genome-scan for susceptibility genes suggests a locus on chromosome 4q24-q27. Eur J Hum Genet 2001; 9: 945–952.
Yokouchi Y, Shibasaki M, Noguchi E et al: A genome-wide linkage analysis of orchard grass-sensitive childhood seasonal allergic rhinitis in Japanese families. Genes Immun 2002; 3: 9–13.
Dizier MH, Bouzigon E, Guilloud-Bataille M et al: Genome screen in the French EGEA study: detection of linked regions shared or not shared by allergic rhinitis and asthma. Genes Immun 2005; 6: 95–102.
Bu LM, Bradley M, Soderhall C, Wahlgren CF, Kockum I, Nordenskjold M : Genome-wide linkage analysis of allergic rhinoconjunctivitis in a Swedish population. Clin Exp Allergy 2006; 36: 204–210.
Barnes KC, Freidhoff LR, Nickel R et al: Dense mapping of chromosome 12q13.12-q23.3 and linkage to asthma and atopy. J Allergy Clin Immunol 1999; 104: 485–491.
Haagerup A, Borglum AD, Binderup HG, Kruse TA : Fine-scale mapping of type I allergy candidate loci suggests central susceptibility genes on chromosomes 3q, 4q and Xp. Allergy 2004; 59: 88–94.
Brasch-Andersen C, Haagerup A, Borglum AD, Vestbo J, Kruse TA : Highly significant linkage to chromosome 3q13.31 for rhinitis and related allergic diseases. J Med Genet 2006; 43: e10.
Christensen U, Moller-Larsen S, Nyegaard M et al: Linkage of atopic dermatitis to chromosomes 4q22, 3p24 and 3q21. Hum Genet 2009.
Haagerup A, Bjerke T, Schiotz PO, Binderup HG, Dahl R, Kruse TA : Asthma and atopy - a total genome scan for susceptibility genes. Allergy 2002; 57: 680–686.
Nyegaard M, Severinsen JE, Als TD et al: Support of association between BRD1 and both schizophrenia and bipolar affective disorder. Am J Med Genet B Neuropsychiatr Genet 2010; 153B: 582–591.
Lange C, DeMeo D, Silverman EK, Weiss ST, Laird NM : PBAT: tools for family-based association studies. Am J Hum Genet 2004; 74: 367–369.
Sleiman PM, Flory J, Imielinski M et al: Variants of DENND1B associated with asthma in children. N Engl J Med 2010; 362: 36–44.
Ott J : Analysis of Human Genetic Linkage, 3rd edn. Baltimore: The John Hopkins University Press, 1999.
Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES : Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 1996; 58: 1347–1363.
Strauch K, Fimmers R, Kurz T, Deichmann KA, Wienker TF, Baur MP : Parametric and nonparametric multipoint linkage analysis with imprinting and two-locus-trait models: application to mite sensitization. Am J Hum Genet 2000; 66: 1945–1957.
Greenberg DA, Berger B : Using lod-score differences to determine mode of inheritance: a simple, robust method even in the presence of heterogeneity and reduced penetrance. Am J Hum Genet 1994; 55: 834–840.
Lander E, Kruglyak L : Genetic dissection of complex traits - guidelines for interpreting and reporting linkage results. Nat Genet 1995; 11: 241–247.
Horvath S, Xu X, Laird NM : The family based association test method: strategies for studying general genotype--phenotype associations. Eur J Hum Genet 2001; 9: 301–306.
Horvath S, Xu X, Lake SL, Silverman EK, Weiss ST, Laird NM : Family-based tests for associating haplotypes with general phenotype data: application to asthma genetics. Genet Epidemiol 2004; 26: 61–69.
Haines JL, Pericak-Vance M : Approaches to Gene Mapping in Comlex Human Diseases. New York: Wiley-Liss, 1998.
Denham S, Koppelman GH, Blakey J et al: Meta-analysis of genome-wide linkage studies of asthma and related traits. Respir Res 2008; 9: 38.
Ober C, Hoffjan S : Asthma genetics 2006: the long and winding road to gene discovery. Genes Immun 2006; 7: 95–100.
Bierbaum S, Nickel R, Koch A et al: Polymorphisms and haplotypes of acid mammalian chitinase are associated with bronchial asthma. Am J Respir Crit Care Med 2005; 172: 1505–1509.
Blumenthal MN, Langefeld CD, Beaty TH et al: A genome-wide search for allergic response (atopy) genes in three ethnic groups: collaborative study on the genetics of asthma. Hum Genet 2004; 114: 157–164.
Rodriguez E, Illig T, Weidinger S : Filaggrin loss-of-function mutations and association with allergic diseases. Pharmacogenomics 2008; 9: 399–413.
Pankratz VS, Iturria SJ : A pedigree partitioning approach to quantitative trait loci mapping of IgE serum level in the GAW12 Hutterite data. Genet Epidemiol 2001; 21 (Suppl 1): S258–S263.
Webb BT, van den Oord E, Akkari A et al: Quantitative linkage genome scan for atopy in a large collection of Caucasian families. Hum Genet 2007; 121: 83–92.
Joki-Erkkila VP, Karjalainen J, Hulkkonen J et al: Allergic rhinitis and polymorphisms of the interleukin 1 gene complex. Ann Allergy Asthma Immunol 2003; 91: 275–279.
Cheng L, Enomoto T, Hirota T et al: Polymorphisms in ADAM33 are associated with allergic rhinitis due to Japanese cedar pollen. Clin Exp Allergy 2004; 34: 1192–1201.
Moller-Larsen S, Nyegaard M, Haagerup A, Vestbo J, Kruse TA, Borglum AD : Association analysis identifies TLR7 and TLR8 as novel risk genes in asthma and related disorders. Thorax 2008; 63: 1064–1069.
Allen M, Heinzmann A, Noguchi E et al: Positional cloning of a novel gene influencing asthma from chromosome 2q14. Nat Genet 2003; 35: 258–263.
Li X, Howard TD, Zheng SL et al: Genome-wide association study of asthma identifies RAD50-IL13 and HLA-DR/DQ regions. J Allergy Clin Immunol 2010; 125: 328–335, e311.
Moffatt MF, Gut IG, Demenais F et al: A large-scale, consortium-based genomewide association study of asthma. N Engl J Med 2010; 363: 1211–1221.
Ober C, Tan Z, Sun Y et al: Effect of variation in CHI3L1 on serum YKL-40 level, risk of asthma, and lung function. N Engl J Med 2008; 358: 1682–1691.
Kawai T, Takeshita S, Imoto Y et al: Associations between decay-accelerating factor polymorphisms and allergic respiratory diseases. Clin Exp Allergy 2009; 39: 1508–1514.
Bottema RW, Kerkhof M, Reijmerink NE et al: X-chromosome Forkhead Box P3 polymorphisms associate with atopy in girls in three Dutch birth cohorts. Allergy 2010; 65: 865–874.
Dietter J, Mattheisen M, Furst R, Ruschendorf F, Wienker TF, Strauch K : Linkage analysis using sex-specific recombination fractions with GENEHUNTER-MODSCORE. Bioinformatics 2007; 23: 64–70.
Acknowledgements
We thank the patients and their families for participating in the project. The helpful and supportive staffs at the outpatient allergy clinics in Viborg, Aarhus, Hjoerring, Herning, and Aalborg and the laboratory technicians in Hjoerring and Aalborg are greatly appreciated. The project was funded by Region Hospital Viborg, the Health Research Fund of Central Denmark Region, the Viborg country health-science research committee, Sygeforsikringen ‘Danmark's healthfund, the Toyota Foundation of Denmark, the A.P. Møller Foundation for the Advancement of Medical Science, the Else and Helene Alstrups Fund, the Lundbeck Foundation, the Villum Kann Rasmussen Foundation, and the Faculty of Health Sciences, Aarhus University.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Kruse, L., Nyegaard, M., Christensen, U. et al. A genome-wide search for linkage to allergic rhinitis in Danish sib-pair families. Eur J Hum Genet 20, 965–972 (2012). https://doi.org/10.1038/ejhg.2012.46
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.46
