
Abstract
Autosomal recessive cerebellar ataxia (ARCA) is a group of neurological disorders characterized by degeneration or abnormal development of the cerebellum and spinal cord. ARCA is clinically and genetically highly heterogeneous, with over 20 genes involved. Exome sequencing of a girl with ARCA from non-consanguineous Dutch parents revealed two pathogenic variants c.37G>C; p.D13H and c.946A>T; p.K316* in CWF19L1, a gene with an unknown function, recently reported to cause ARCA in a Turkish family. Sanger sequencing showed that the c.37G>C variant was inherited from the father and the c.946A>T variant from the mother. Pathogenicity was based on the damaging effect on protein function as the c.37G>C variant changed the highly conserved, negatively charged aspartic acid to the positively charged histidine and the c.946A>T variant introduced a premature stop codon. In addition, 27 patients with ARCA were tested for pathogenic variants in CWF19L1, however, no pathogenic variants were identified. Our data confirm CWF19L1 as a novel but rare gene causing ARCA.
Similar content being viewed by others
Introduction
Autosomal recessive cerebellar ataxia (ARCA) is a group of rare, genetically and clinically heterogeneous neurological disorders affecting both the peripheral and central nervous systems, mainly occurring in children and young adults.1 Clinical manifestations range from progressive cerebellar syndromes, as a key feature, to spinocerebellar symptoms with spasticity, ophthalmoplegia, sensory/motor neuropathy, involuntary movements, seizures, cognitive impairment, skeletal abnormalities and cutaneous disorders. To date, more than 20 genes have been reported to cause ARCA, however, explaining only 40% of cases.2 Recently, pathogenic variants were described in a novel gene (CWF19L1) in two siblings with autosomal recessive ataxia syndrome.3 Here, we report a second case, a girl from non-consanguineous Dutch parents, with cerebellar ataxia and atrophy, who was compound heterozygous for pathogenic variants in the CWF19L1 gene. Our results confirm the suggested role of CWF19L1 in rare cases of autosomal recessive cerebellar ataxia.
Materials and methods
Index patient and 27 additional ARCA patients
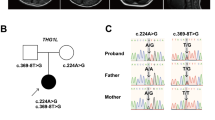
The index patient was born from non-consanguineous parents (Figure 1a) at term after an uncomplicated delivery and had a birth weight of 2480 g. During pregnancy, the echoes of the head showed a height comparable to the second percentile within the Dutch population. Complaints of clumsiness were noted at 5 years of age. In addition, apart from being a slow learner, there were complaints of mild weakness and fatigability. Neurological investigation at the age of 10 showed a height of 126 cm (P1), weight of 24 kg (weight-length P50) and skull circumference of 53 cm (P50). Furthermore, she showed a slurred, monotonic, sometimes even staccato-like speech and, although she was cooperative, slow in her performance. There was an oculomotor apraxia, no nystagmus and a status after surgical strabismus correction. No retinal pigment layer alterations were noticed and no seizures reported. The patient had normal strength, but a slight dystonic movement disorder, particularly in the arms. A mild hypotonia with unsteady stance and broad-based gait was observed as well as intention tremor and overshoot with the finger–nose test. In the tandem walking test, she was more than 5 cm off, with an increase in unsteadiness over a period of 1 year. There were no signs of a sensory ataxia or polyneuropathy and no pyramidal tract signs with symmetric and normal reflexes. Family history did not reveal any neurological abnormalities.

MRI and exome data of the index patient. (a) Pedigree of a Dutch family with autosomal recessive cerebellar ataxia and atrophy. The affected subject is indicated in black. CWF19L1 variants are indicated, illustrating that the c.37G>C is inherited from the father and c.946A>T from the mother. (b) The midline sagittal T1-weighted brain magnetic resonance imaging (MRI) demonstrating at the age of 9 years a severe vermian cerebellar hypoplasia with prominent fissures and prominent surrounding cisterns (b-1). Axial T2-weighted MRI showing a slight aspecific enlargement of the ventricles and cerebellar hypoplasia (b-2 and b-3). (c) Schematic representation of the filtering steps of the whole-exome data.
Extensive metabolic workup in blood and urine did not reveal any abnormalities. In cerebrospinal fluid, there was an increase detected in myelin basic protein (MBP): 1.2 μg/l (normal: 0.3–1.0 μg/l), whereas concentrations of GFAP, S100 and tau-protein were normal. Magnetic resonance imaging of the brain showed a severe cerebellar hypoplasia, particularly of the vermis (Figure 1b). Compared with 5 years earlier, there was a slight decrease detected in the cerebellar vermis and hemisphere volume.
An additional 27 patients with genetically undiagnosed ARCA were selected based on ataxia as main clinical symptom and confirmed by cerebellar hypoplasia demonstrated by MRI imaging.
Whole-exome sequencing and sequence analysis in CWF19L1
After DNA extraction, the exome of the index patient was captured using an exome enrichment kit including the UTR regions (Agilent SureSelect version 4) according to the manufacturers’ protocol. Sequencing was performed on an Illumina HiSeq2000 using a 2x100 bp paired-end recipe. Basecalling and demultiplexing was done using bcl2fastq 1.8.4, reads were aligned onto the human reference genome (hg19) using BWA 0.5.9,4, 5 duplicates marked using the PICARD software suite 1.77 (http://picard.sourceforge.net) and variants were called using GATK 2.1–8.6, 7 Annotations were added using an in-house-built annotation database, according to the UCSC RefGene track, dbSNP137 and the dbNSFP (v2.0).8 We filtered for genes that contained at least two nonsynonymous variants, variants leading to a premature stop codon or splice variants with allele frequencies of <0.01 in dbSNP137 and absence in our in-house database of ~200 exomes. The variants located in these genes were further evaluated for the impact on protein function by SIFT and Polyphen2 as predicted by the dbNSFP database.
Pathogenic variants identified in CWF19L1 and phenotypes of the index patient were submitted to the gene variant database http://databases.lovd.nl (patient ID 00037591),
For the identification of pathogenic variants in CWF19L1 in additional patients with cerebellar ataxia and atrophy, the exons and flanking introns of CWF19L1 were amplified with specific intronic primers (sequences available on request).
RNA extraction and real-time PCR
Human fibroblasts were cultured in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% L-glutamine and 1% penicillin/streptomycin (Gibco BRL, Grand Island, NY, USA), and incubated in humidified atmosphere of 5% CO2 at 37 °C. Total RNA was extracted with TRIzol (Invitrogen, Carlsbad, CA, USA) and converted into cDNA using RT First Strand Kit (Qiagen, Redwood City, CA, USA). Expression of CWF19L1 was determined using SYBR Green qPCR on a 7900HT RT-PCR System (Life Technologies, Carlsbad, CA, USA). TBP was used to normalize expression levels.
To test for nonsense-mediated decay, PCR amplification of cDNA with primers designed to flank the variant introducing the premature stop codon is followed by Sanger sequencing.
Results
Exome sequencing was performed in a patient with ARCA, from Dutch non-consanguineous parents, to identify the underlying genetic defect (Figure 1). In total, 52,190 variants were detected. After removing intergenic UTR-variants, non-splice site-related intronic variants, synonymous variants, variants with an allele frequency >0.01 in dbSNP137 and variants present in our in-house database, only 253 variants were left. Seven genes harbored ≥2 variants of which only two (CWF19L1 and MYO18B) harbored two variants predicted to be damaging according to SIFT (score <0.05) and Polyphen2 (score >0.85) or resulted in a premature stop codon. MYO18B is unlikely to cause ARCA as variants in this gene are associated with lung cancer9 or linked to Klippel–Feil anomaly and myopathy,10 which were not present in our patient. However, during the course of our studies, CWF19L1 was reported as a novel cause of ARCA3 and we focused further on this gene. The CWF19L1 c.37G>C (r.37G>C) variant results in the substitution of the highly conserved negatively charged aspartic acid to the positively charged histidine at amino acid position 13 (Figure 2a), whereas the c.946A>T (r.946A>U) substitution results in a premature stop codon at amino acid position 316, (NM_018294.4; Figure 2a).

(a) Schematic representation of CWF19L1 and positions of variants. (b) Conservation of affected amino acid residue of p.Asp13His from H. sapiens to C. elegans (affected amino acid shown in bold).
Sanger sequencing showed that the father was heterozygous for the c.37G>C variant and the mother for c.946A>T variant, confirming that the patient was compound heterozygous (Figure 1a).
CWF19L1 expression level was normal in the patient’s fibroblasts compared with control (Figure 3a). Sequence analysis of cDNA from the patient’s fibroblasts showed a decreased amount of the r.946A>U transcript compared with the r.37G>C (Figure 3b), which was not seen on DNA level, indicating this allele is degraded by nonsense-mediated mRNA decay.

CWF19L1 mRNA expression and maturation. (a) CWF19L1 expression in the patient’s fibroblasts. Error bars indicate standard deviation. (b) Sequence of CWF19L1 cDNA product shows decreased amount of the c.946A>T transcript compared with the c.37G>C transcript.
Subsequently, 27 additional patients with autosomal recessive cerebellar ataxia and atrophy were selected and screened for variants in CWF19L1. However, no pathogenic variants were identified in CWF19L1 for these patients.
Discussion
Using whole-exome sequencing, we identified two novel heterozygous variants in CWF19L1 causing ARCA in a Dutch non-consanguineous family. One variant p.D13H changes the highly conserved, negatively charged aspartic acid into the positively charged histidine and is predicted to affect protein stability or function. The other variant leads to a premature stop codon p.K316* which results in nonsense-mediated mRNA decay. Total mRNA of the patient’s fibroblasts was expressed at a level similar to the control indicating that increased c.37G>C transcript compensated the degradation by nonsense-mediated decay.
During the course of our studies, CWF19L1 was recently suggested as a novel ataxia gene in a Turkish family with a comparable phenotype to our patient (Table 1).3 The authors showed that knockdown of the homolog of CWF19L1 in zebrafish results in cerebellar and movement abnormalities and that the encoded protein C19L1 is expressed throughout the brain suggesting an important role for C19L1 in neural development.3 Although the exact function of the C19L1 is still unknown, the nuclear localization in combination with a metallophosphatase domain, which is found in RNA lariat debranching enzymes, suggests a role in mRNA processing.3 However, further studies are needed to determine the exact function of this protein and its role in brain development. Detailed functional characterization of C19L1 will provide more information on different causes of ARCA and possibly reveal novel candidates for the analysis of pathogenic variants in patients and/or targets for therapy. Screening of CWF19L1 for pathogenic variants in an additional 27 genetically undiagnosed patients with ARCA did not reveal any additional pathogenic variants, indicating variants in CWF19L1 are a rare cause of ARCA.
In conclusion, our report confirms the recent study on pathogenic variants in CWF19L1 as a possible cause of ARCA, adding CWF19L1 to the already long list of candidate genes for patients with ARCA.
References
Anheim M, Tranchant C, Koenig M : The autosomal recessive cerebellar ataxias. N Engl J Med 2012; 366: 636–646.
Sailer A, Houlden H : Recent advances in the genetics of cerebellar ataxias. Curr Neurol Neurosci Rep 2012; 12: 227–236.
Burns R, Majczenko K, Xu J et al: Homozygous splice mutation in CWF19L1 in a Turkish family with recessive ataxia syndrome. Neurology 2014; 83: 2175–2182.
Li H, Durbin R : Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–1760.
Li H, Durbin R : Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010; 26: 589–595.
McKenna A, Hanna M, Banks E et al: The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–1303.
DePristo MA, Banks E, Poplin R et al: A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011; 43: 491–498.
Liu X, Jian X, Boerwinkle E : dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mutat 2013; 34: E2393–E2402.
Nishioka M, Kohno T, Tani M : MYO18B, a candidate tumor suppressor gene at chromosome 22q12.1, deleted, mutated, and methylated in human lung cancer. Proc Natl Acad Sci USA 2002; 99: 12269–12274.
Alazami AM, Kentab AY, Fageih E et al: A novel syndrome of Klippel-Feil anomaly, myopathy, and characteristic facies is linked to a null mutation in MYO18B. J Med Genet 2015; 52: 400–404.
Acknowledgements
This work is supported by the Alma in Silico project, which is financed by the Interreg IV European funds, The Walloon Region, The North Rhine Westphalia, The Flemish Community, The Belgian Province of Limburg and The Dutch Province of Limburg, as well as by the Universities of Maastricht and Liège, by the Prinses Beatrix Spierfonds (grant W.OR11-24) and the Stichting Metakids.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Nguyen, M., Boesten, I., Hellebrekers, D. et al. Pathogenic CWF19L1 variants as a novel cause of autosomal recessive cerebellar ataxia and atrophy. Eur J Hum Genet 24, 619–622 (2016). https://doi.org/10.1038/ejhg.2015.158
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.158
This article is cited by
-
RIP-PEN-seq identifies a class of kink-turn RNAs as splicing regulators
Nature Biotechnology (2024)
-
Novel CWF19L1 mutations in patients with spinocerebellar ataxia, autosomal recessive 17
Journal of Human Genetics (2023)
-
Pathogenic SLIRP variants as a novel cause of autosomal recessive mitochondrial encephalomyopathy with complex I and IV deficiency
European Journal of Human Genetics (2021)
-
The Classification of Autosomal Recessive Cerebellar Ataxias: a Consensus Statement from the Society for Research on the Cerebellum and Ataxias Task Force
The Cerebellum (2019)
-
Novel candidate genes and variants underlying autosomal recessive neurodevelopmental disorders with intellectual disability
Human Genetics (2018)
