
Abstract
Fragile X syndrome is the most common heritable form of intellectual disability and is caused by the expansion over 200 repeats and subsequent methylation of the CGG triplets at the 5′ UTR of the FMR1 gene, leading to its silencing. The epigenetic and molecular mechanisms responsible for FMR1 gene silencing are not fully clarified. To identify structure-specific proteins that could recruit components of the silencing machinery we investigated the role of CGGBP1 in FMR1 gene transcription. CGGBP1 is a highly conserved protein that binds specifically to unmethylated CGG tracts. Its role on FMR1 transcription is yet to be defined. Sequencing analysis and expression studies through quantitative PCR of CGGBP1 were performed in cell lines with different allele expansions: wild type, premutation, methylated full mutation and unmethylated full mutation, demonstrating no differences between them. ChIP assays clearly demonstrated that CGGBP1 binds to unmethylated CGG triplets of the FMR1 gene, but not to methylated CGGs. We also observed that CGGBP1 binding to the FMR1 locus was restored after pharmacological demethylation, with 5-azadC, of alleles, carriers of methylated full mutation, suggesting a possible role for CGGBP1 in FMR1 expression. CGGBP1 silencing with shRNAs (reaching ~98% of CGGBP1-mRNA depletion) did not affect FMR1 transcription and CGG expansion stability in expanded alleles. Although the strong binding to the CGG tract could suggest a relevant role of CGGBP1 on FMR1 gene expression, our results demonstrate that CGGBP1 has no direct effect on FMR1 transcription and CGG repeat stability.
Similar content being viewed by others

Introduction
Fragile X syndrome (FXS, OMIM #300624) is the leading cause of heritable intellectual disability and autism, belonging to the group of the so-called FRAXopathies.1, 2 FXS affects ~1 in 4000 males and 1 in 9000 females.3, 4 It is almost invariably due to a large expansion (full mutation, FM) of an instable CGG repeat in the 5′ untranslated region (5′-UTR) of the FMR1 gene (in Xq27.3). CGG expansion over 200 repeats (FM) is followed by epigenetic modifications of the 5′-UTR of the FMR1 gene, including primarily DNA methylation in both the expanded CGG tract and in the neighboring CpG island, as well as other heterochromatic histone modifications (ie, methylation of H3K9, trimethylation of H3K27, H3 and H4 hypoacetylation). These epigenetic modifications cause the transcriptional silencing of the FMR1 gene and consequently the lack of the FMRP protein.5, 6, 7 FMRP is an RNA-binding protein that is involved in multiple pathways of mRNA metabolism, essentially as an inhibitor of translation particularly at synaptic level.8 Several years ago have been described rare individuals of normal intelligence, carrying a transcriptionally active unmethylated full mutation (UFM).9 The epigenetic characterization of these rare UFM cell lines showed histone marks similar to euchromatin with absence of DNA methylation in both the CpG island of the promoter region and the CGG repeat itself. Levels of FMR1 transcription are slightly increased compared with controls, but translational efficiency of the corresponding mRNA is reduced.6, 7 These studies demonstrated that CGG expansion in the range of FM per se does not prevent FMR1 transcription, DNA methylation being the necessary further step. The mechanisms leading to lack of DNA methylation, however, remain to be elucidated. During the early stages of development full mutations are unmethylated and the FMR1 gene is active.10 Methylation and gene silencing occur at about 11 weeks of gestation.11 One could speculate that an enzyme or a molecular pathway, not yet identified, involved in the establishment of DNA methylation pattern is inactive/inefficient in the rare carriers of an unmethylated full mutation.
Once methylation is established several other proteins specifically bind to the unmethylated or methylated CGG tract ensuring stability of the repeat tract and its methylation status throughout cell division.12 CGG repeat stability has a crucial role in the pathogenesis of FXS. Intrinsic properties in the CGG expansion are the major elements capable of stabilizing the repeats. The periodic AGG ‘interruptions’ or ‘anchors’ within the polymorphic CGG sequence prevent expansion from intermediate/small premutation alleles to FM and maintain the correct functional organization of the FMR1 gene in genomic packaging.13, 14 Recent reports show that ancillary molecules of synthetic nature can further stabilize the secondary structures that are formed from a high number of CGG repeats.15 A good candidate for this role is the CGGBP1 protein. First described by Deissler et al, the CGGBP1 protein (also known p20) is a 20-kDa nuclear protein that binds specifically CGG repeat tracts. It is coded by a gene localized on chromosome 3p11.1 and consists of four exons spanning 19 839 bp.16 CGGBP1 is a 167-aa protein with at least one DNA-binding motif and a nuclear localization signal. The CGGBP1 gene has a CpG island in its promoter region, which is completely unmethylated in human cells.17 It is highly conserved among mammals and it is expressed in a variety of human tissues implying an important function in mammalian cells.16 The binding to DNA requires a minimum of eight CGG repeats and is methylation sensitive. Functional studies showed that CGGBP1 is an important protein in correcting cell cycle errors and consequent aging of the cells.18, 19, 20 Despite its ability to bind to CGG tracts, its functional role in FMR1 gene transcription is poorly known. In co-transfection experiments it was demonstrated that CGGBP1 reduces the FMR1 promoter activity in HeLa cells.21 Sequencing analysis also showed that CGGBP1 gene is unmodified in the genome of normal, premutated and FXS individuals.21
For the first time we studied the role of CGGBP1 protein in FXS cell lines to address its role both in transcriptional regulation and in repeat stability of several classes of FMR1 alleles. We performed sequencing analysis of the CGGBP1 gene and quantification of its transcript in cell lines derived from normal, premutation, FXS and UFM individuals. We confirmed that CGGBP1 binds to the CGG tract in the 5′-UTR of the FMR1 gene and that this binding is clearly methylation sensitive. CGGBP1 depletion with shRNAs apparently did not affect either FMR1 transcription activity in transcriptionally active alleles or CGG expansion stability in expanded alleles. Hence, although a strong binding of CGGBP1 protein to the CGG tract of the FMR1 gene was demonstrated, our results showed that CGGBP1 is not a direct regulator of FMR1 transcription.
Materials and Methods
Cell lines and pharmacological treatments
Lymphoblastoid cell lines were established by Epstein–Barr virus transformation from peripheral blood lymphocytes of two PM (premutations: PM1 and PM2), three FXS (methylated full mutation: FXS1, FXS2 and FXS3), one UFM (unmethylated full mutation) and two normal control (WT1 and WT2) males. FXS3 is a mosaic for size and methylation (Supplementary Figure S1 on Supplementary Information). Lymphoblasts were grown in RPMI1640 medium (Sigma-Aldrich, Milan, Italy; R0883) supplemented with 20% fetal bovine serum, 2.5% l-glutamine and 1% penicillin/streptomycin at 37 °C with 5% CO2. FXS lymphoblasts were treated twice with the demethylating agent 5-aza-2′-deoxycytidine (5-azadC) (Sigma-Aldrich, A3656) at 1 μm final concentration for 8 consecutive days, as previously described.22 At the end of the treatments, cells were harvested, cell viability was measured by the propidium iodide method (Nucleocounter, Sartorius/Stedim, Florence, Italy) and nucleic acids were extracted to check for FMR1 reactivation, DNA methylation (see Supplementary Information) and chromatin structure. Primary fibroblast cultures were obtained from skin biopsies derived from UFM individual. Fibroblasts from WT and FXS were provided by the Coriell Institute (Camden, NJ, USA). Fibroblasts were grown in DMEM complete medium (Gibco Life Technologies, Carlsbad, CA, USA; 41965-062) at 37 °C with 5% CO2. Packaging cell lines 293LTV (Cell Biolabs, Inc., San Diego, CA, USA; LTV100) for lentiviral production were cultured in DMEM complete medium (Gibco Life Technologies, 41965-062) at 37 °C with 5% CO2.
CGGBP1 gene sequencing
Sequencing analysis of the CGGBP1-coding region was performed on genomic DNA, extracted from WT, FXS and UFM lymphoblastoid cell lines with phenol/chloroform standard method. Primers were designed based on the genomic sequence AF094481.1 of GenBank; the list of primers and PCR conditions are available on request. Sequencing reaction was performed using BigDye terminator v3.1 Cycle Sequencing Kit (Life Technologies) and then were purified with BigDyeXTerminator Purification kit (Life Technologies), according to the manufacturer’s protocol, and processed in 3130 Genetic Analyzer (Life Technologies).
Quantitative RT-PCR analysis
Total RNA was extracted with standard TRIzol reagent protocol (Ambion Life Technologies, Carlsbad, CA, USA; 15596-018). Afterwards, 1 μg of total RNA was retro-transcribed into cDNA by M-MLV-Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA; AM2044) using random hexamers. For a relative quantification of each transcript, the following pre-developed TaqMan assays (Life Technologies) were used: CGGBP1 (Hs00186380_m1), GAPDH (402869) and FMR1 (Hs00233632_m1). The real-time RT-PCR was performed on an ABI7900HT (Life Technologies). Relative quantification of target transcripts was obtained as follows: 2−(ΔCt(target)−ΔCt(control))=2−ΔΔCt, where ΔCt is the difference Ct(target)–Ct(GAPDH), and Ct is the threshold cell cycle in the PCR.
Chromatin immunoprecipitation and quantification of immunoprecipitated DNA
Chromatin immunoprecipitation (ChIP) assay was performed as previously described.23 Binding of CGGBP1 protein was assayed using the specific antibody (Sigma-Aldrich, WH0008545M2). Immunoprecipitated DNA (IP-DNA) was extracted by standard procedure (phenol/chloroform/isoamyl alcohol 25:24:1) and then quantified by real-time PCR (ABI7900HT, Life Technologies) using fluorescent probes and primers specific for promoter, exon 1 and exon 16 of the FMR1 gene, for the HPRT gene (negative control for binding) and for ribosomal subunit 28S gene (positive control for binding) (sequences available on request). Three independent immunoprecipitation experiments were performed in WT, PM, FXS and UFM lymphoblastoid cell lines. CGGBP1 binding was assessed by ChIP also in FXS1 and FXS2 lymphoblastoid lines after 5-azadC treatments.
All variables were analyzed by means of descriptive statistics (mean, median, SD and SEM). ChIP data were expressed as mean of absolute quantity of IP-DNA and analyzed with Student’s t-test. The level of significance was set at P≤0.05. Data analysis was performed using STATA Intercooled v. 9.2 software (Stata Co.; College Station, Lakewag, TX, USA).
Knockdown of CGGBP1 transcript
Knockdown of CGGBP1 transcript was carried out in WT, FXS and UFM fibroblasts using both transient and stable approaches, as detailed in the Supplementary Information.
Western blot analysis
Proteins extracted from untreated and shRNA-treated WT and UFM fibroblasts were resuspended in Laemli buffer, boiled, separated on 10% polyacrylamide gel electrophoresis, transferred to Hybond-ECL membrane (GE Healthcare, Milan, Italy) and visualized with a transilluminator (Thermo-Fisher, Milan, Italy), using the ECL Western Blotting Detection Reagents (GE Healthcare), according to the manufacturer. Primary antibodies were used at the following concentrations: 1:1000 anti-CGGBP1 mouse antibody (Santa-Cruz Biotechnology, Paso Robles, TX, USA), 1:1000 anti-FMRP mouse policlonal antibody (Immunological Science, Rome, Italy) and 1:10000 anti-GAPDH mouse antibody (Sigma-Aldrich).
CGG expansion analysis through fluorescent PCR
The analysis of CGG-tract expansion in FXS and UFM cell lines was performed using fluorescent primers and GC-rich PCR system (Roche, Monza, Italy). The PCR products were then separated on capillary electrophoresis on 3130 Genetic Analyzer (Life Technologies) and the result was analyzed with Sequencing Analysis v5.2 software (Life Technologies).
Results
Sequencing analysis and expression levels of CGGBP1 reveal no variations in cell lines carrying different FMR1 alleles
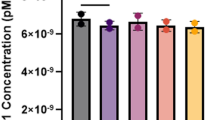
We first sequenced the CGGBP1 gene in genomic DNA extracted from WT, PM, FXS and UFM lymphoblastoid cell lines. We covered the entire coding region of the CGGBP1 gene, including its 3′-UTR, and confirmed the presence of a third exon, which is typically human, in all cell lines analyzed. Sequence analysis showed no significant variants in any of the cell lines (data not shown). Given the absence of variants in the coding sequence of CGGBP1 gene, we also tested the basal transcription of CGGBP1, demonstrating an almost equal expression level in all cell lines analyzed (WT, PM, FXS and UFM) both lymphoblasts and fibroblasts (Supplementary Figures S2A and S2B, respectively).
CGGBP1 binds unmethylated fully mutated FMR1 gene
We proceeded by analyzing CGGBP1 binding to the FMR1 gene. The results of ChIP analysis are reported as absolute DNA values in Figure 1. We investigated three regions of the FMR1 gene, the promoter (Figure 1a), exon 1 (Figure 1b) and exon 16 (Figure 1c). This latter region may be considered as a negative control for CGGBP1 binding as it is far from the CGG sequence. Moreover, we tested the HPRT gene and the ribosomal subunit 28S gene, as negative and positive controls, respectively (Figures 1d and e). The promoter of the 28S gene is CGG rich, while the HPRT gene does not have repetitive tracts. ChIP assays were performed in two different WT, three FXS, two PM and one UFM lymphoblastoid cell lines. Three independent immunoprecipitations demonstrated that generally the CGGBP1 protein is significantly more abundant in the exon 1 compared with the promoter region in all transcriptionally active alleles analyzed (PM and UFM). On the other hand, UFM cells demonstrated significantly higher amounts of CGGBP1 bound compared with WT, on both promoter and exon 1. As expected, CGGBP1 binding to FMR1 in FXS cells is significantly lower compared with the other cell lines. The mosaic cell line (FXS3) demonstrated a CGGBP1-binding profile similar to the two FXS cell lines, despite the presence of an unmethylated amplification within the premutation range. The exon 16 region showed the lowest level of CGGBP1 binding, even when compared with the negative control gene, HPRT. These results indicate that the CGGBP1 protein strongly binds to transcriptionally active and unmethylated FMR1 alleles.

ChIP assays for CGGBP1 binding to the FMR1 gene. Three different immunoprecipitations (n=3) were set up to test the binding of CGGBP1 protein on the FMR1 locus in two normal lymphoblastoid cell lines (WT1 and WT2), three FXS (FXS1, FXS2 and FXS3, this latter being a mosaic for size and methylation), two PM (PM1 and PM2) and one UFM carrier. The FMR1 regions investigated are the promoter (a), exon 1 (b) and exon 16 (c) (the last does not contain CGG repeats and can be considered as a negative control for CGGBP1 binding). HPRT gene (d) and 28S promoter (e) were used as negative and positive controls for CGGBP1 binding, respectively. The quantities reported on the y axis are absolute values of IP-DNA expressed in nanograms with respective standard error bars. In transcriptionally active cell lines (PM and UFM) the levels of CGGBP1 binding are significantly higher with respect to normal controls (WT), particularly in the promoter and exon 1 region of the FMR1 gene. The asterisks (*) indicate the values that reached statistical significance (P<0.05). The differences between WT, FXS, PM and UFM in exon 16, HPRT gene and 28S promoter do not reach statistical significance.
CGGBP1 binding to FMR1 is restored after pharmacological demethylation
The presence, at best at low levels, of CGGBP1 in the FXS alleles prompted us to explore its binding after pharmacological demethylation of the FMR1 gene. We treated twice two different FXS lymphoblastoid cell lines (FXS1 with around 250 CGGs and FXS2 with 350 CGGs) with the demethylating agent 5-azadC for 8 days at the final concentration of 1 μm, as previously reported.22 As observed in our previous works,22, 23, 24 the demethylating treatment with 5-azadC was able both to restore the FMR1 transcription, at levels of FMR1–mRNA that were 35% (FXS1) and 25% (FXS2) of control, respectively, and to demethylate its promoter (Supplementary Figure S3). ChIP assays for CGGBP1 protein performed after the demethylating treatment demonstrated that the binding of CGGBP1 to the promoter and the exon 1 regions was restored (Figure 2). In fact, the binding of CGGBP1 to the FXS1 expanded allele was higher than to the WT alleles. This effect was more marked in FXS1 compared with FXS2. The variability in the two cell lines was probably due to the difference in CGG expansion. The FXS2 cell line has a longer CGG repeat sequence compared with FXS1, and can be only partially demethylated. For this same reason the increase in transcription reactivation and CGGBP1 binding to promoter and exon 1 regions are higher in the FXS1 cell line compared with FXS2 line. Negative controls (exon 16 of FMR1 gene and HPRT gene) did not undergo a significant change of the CGGBP1-binding profile after 5-azadC treatment, while a positive control (28S promoter) was shown to bind more CGGBP1 after 5-azadC, particularly in FXS2, probably due to a higher level of demethylation in this region in FXS2 compared with FXS1. These data confirm that the binding of CGGBP1 to FMR1 is methylation sensitive and can be restored by 5-azadC-induced demethylation.

ChIP assay for the CGGBP1 to the FMR1 locus after pharmacological demethylation with 5-azadC in FXS cell lines. Two FXS lymphoblastoid cell lines were treated twice with 5-azadC at 1 μm (final concentration) for 8 days to obtain a demethylation of the regulatory region of the FMR1 gene: FXS1 (a) and FXS2 (b), with ~250 and 350 CGGs, respectively. Transcriptional reactivation (Supplementary Figure S3) in both cell lines is a clear indication of at least partial demethylation of the region. In fact CGGBP1 binding is restored in the promoter and exon 1 of the FMR1 gene after 8 days of treatment in both cell lines and in both independent experiments (5-azadC1 and 2). The values represent absolute quantities of IP-DNA expressed in nanograms with respective standard error bars. The asterisks (*) indicate the differences that reached statistical significance (P<0.05).
CGGBP1 depletion does not affect FMR1 transcription
Given the CGGBP1 binding to unmethylated and pharmacologically demethylated FMR1 gene, we went on to check the functional role of CGGBP1 on FMR1 transcriptional regulation. As CGGBP1 knockout mice are not available (http://jaxmice.jax.org/index.html) and the literature is poor of information about the viability of cell lines defective for CGGBP1, we established transient and stable CGGBP1 knockdown cell lines with shRNAs through lentiviral infection. In both transfection experiments we noted high rate of cell mortality, possibly due to a role of CGGBP1 in regulating cell cycle progression and apoptosis. Transient transfections were performed with both esiRNA and shRNA against CGGBP1 transcript. Knockdown of CGGBP1 mRNA with esiRNA (Supplementary Figure S4) showed a reduction of around 85–90% of CGGBP1 transcript in both WT and UFM cell lines after 72 h without significant changes of FMR1 transcription.
Packaging cells 293LTV specifically engineered for lentivirus production were transfected with plasmids needed for both virus production (psPAX2 and pMD2.G) and specific shRNAs directed against CGGBP1 transcript. We tested five different shRNA clones (listed in Supplementary Table S1 in the Supplementary Information). Already at 24 h from infection, the cells transfected with the GFP reporter protein were becoming fluorescent. After 48 h, GFP expression was present in essentially all cells. At subsequent time points the reporter protein continued to be detectable (data not shown). Two independent transient co-transfections with shRNA 4 and 5 proved that the residual levels of CGGBP1 transcription were around 1–2% compared with untreated controls both in WT and in UFM fibroblasts (Figures 3a and b, left panels). Infection with empty vector (mock) did not change the levels of CGGBP1 transcription. Despite the high degree of CGGBP1-mRNA silencing, FMR1 transcription was either unchanged or slightly higher compared with the untreated WT and UFM controls (Figures 3a and b, right panels). Even in transient transfections of FXS fibroblasts CGGBP1-mRNA level was reduced to about 5% of the untreated controls, again with unmodified FMR1 gene transcription levels (Supplementary Figure S5). The efficiency of CGGBP1 transient depletion was also confirmed by the low expression of the CGGBP1 protein (Figure 3c), while FMRP levels remained unmodified after CGGBP1 knockdown in both WT and UFM, as expected (Figure 3d).

Relative quantification of CGGBP1 and FMR1 transcripts by fluorescent real-time RT-PCR after transient transfection with shRNAs against CGGBP1 mRNA. The silencing levels of CGGBP1 mRNA (left panels) are very high, reaching 98% of knockdown (shRNA 4-5 in combination) compared with the untreated control both in WT (a) and in UFM (b). Levels of FMR1–mRNA (right panels) does not significantly changes after CGGBP1 silencing in both cell line types. These results were confirmed also by western blot (c), in which a very low residual CGGBP1 protein was detected after mRNA depletion. FMRP protein levels remained unchanged after CGGBP1 silencing in both WT and UFM (d).
Stable depletion of CGGBP1 does not affect CGG expansion stability
All cell lines transfected with lentiviral shRNAs were put under antibiotic selection. We were able to isolate only few single-cell colonies that showed an extremely slow replication rate. Previous mortality curves showed that the concentration of puromycin (1.5 μm) was adequate (data not shown). After 20 days from antibiotic selection, RNA and DNA extractions were performed. CGGBP1 transcript silencing and FMR1 expression were comparable to those observed in transient transfections (Figure 4a).

Relative quantification of CGGBP1 and FMR1 transcripts by fluorescent real-time RT-PCR after stable transfection with shRNAs against CGGBP1 mRNA and amplification of the CGG expansion. The silencing levels of CGGBP1 mRNA (a) are very high, reaching 98–99% of knockdown particularly combining shRNA 4 and 5 respect to the untreated control in WT, FXS and UFM. Levels of FMR1–mRNA do not change after CGGBP1 silencing in all cell line types. Fluorescent amplification of the CGG tract was performed in FXS (b) and in UFM (c) fibroblasts stable transfected with shRNA 4–5 before and after 20 days of puromycin selection. The CGG expansion in FXS and UFM cell lines remained stable at ~440 and 250 repeats, respectively.
To explain the strong binding of CGGBP1 on the 5′-UTR region of the FMR1 gene, we tested whether its stable removal could affect CGG repeat stability, particularly in expanded cells (FXS and UFM). The stable shRNA-transfected cells and their controls were collected and genomic DNA was extracted. To analyze the stability of the CGG expansion after an extended deprivation of CGGBP1 protein, a fluorescent PCR-based amplification of the CGG repeat was performed. The CGG expansion in the 5′-UTR region of the FMR1 gene remained completely stable after the CGGBP1 depletion in both FXS (Figure 4b) and in UFM (Figure 4c) cells without any sign of mosaicism. Therefore, CGGBP1 binding on FMR1 apparently did not affect CGG expansion stability even after 20 days of CGGBP1 stable absence.
Discussion
Even though the molecular processes involved in FMR1 silencing have been studied for more than two decades and partly clarified, the mechanism responsible for FMR1 silencing has yet to be fully clarified. The issue is particularly complex because two separate events concur in blocking FMR1 transcription, neither of which is fully understood: the expansion of the CGG repeat tract through successive generations and the consequent methylation of cytosines. Currently, it is not even known whether repeat expansion and DNA methylation are sequential events. Although the mechanism of repeat expansion remains uncertain, in general, repeat instability is often due to the formation of alternative DNA secondary/tertiary structures in a parent or a daughter strand during DNA synthesis.25 However, embryonic stem cells with a fully expanded CGG tract maintain FMR1 transcription until differentiation takes place and the first marker that indicates a future silencing of the gene is not DNA methylation, but rather hypermethylation of H3K9.10 In this scenario, individuals with rare UFM perfectly fit as representative of a ‘frozen’ intermediate state of the transition between the expansion of the CGG sequence and its methylation.6, 7 It has recently been shown that during the early stages of cellular differentiation, FMR1 mRNA with expanded CGG triplets over 200 repeats interact with the complementary portion on the DNA sequence.26 The presence of this mRNA–DNA heteroduplex would seem to induce transcriptional silencing through methylation of both the CGG tract and the FMR1 promoter. However, this explanation does not account for the rare individuals with UFM. The existence of UFM indicates that FMR1 gene transcription is possible even in the presence of CGG expansion in the full mutation range (>200 CGGs). In this context, the investigation of DNA-binding proteins associated with FMR1, and in particular with the CGG repeats, may give an important contribution to understanding of the molecular mechanisms that regulate FMR1 transcription.
CGGBP1 could be one of many factors that affects this complex mechanism. It is logically to predict an interaction between this protein and the FMR1 gene, even though the role of CGGBP1 in FMR1 gene regulation has not been investigated in normal or patient cell lines, most work having been done on HeLa or HEK293 cell lines.16, 17, 18, 19, 20, 21
Therefore, we decided to investigate the role of CGGBP1 in FMR1 regulation in a more systematic manner. First, a screening of variants in CGGBP1 gene performed in multiple cell lines with normal, premutated, fully methylated and fully unmethylated mutations of the FMR1 gene demonstrated that none of these cell lines has CGGBP1 genomic variants. Müller-Hartmann et al21 obtained similar results, but they has not tested UFM cell line. Our work allows to rule out the possibility that CGGBP1 variation that modifies the interaction of its protein product with the FMR1 gene can prevent FMR1 methylation in UFM cells. Moreover, no known disease has ever been associated with absence or functional alteration of CGGBP1. To verify whether altered CGGBP1-mRNA expression could be responsible for the unmethylated status, a real-time PCR was performed to define the CGGBP1 expression in the various cell lines. CGGBP1 mRNA was relatively stable in all cell lines tested, with minimal fluctuations that could be observed within the same type of cells (one WT cell line has 1.5 times the amount of the other). These fluctuations in WT cell lines were previously observed for other transcripts, including those of FMR1 mRNA.7 After this, CGGBP1 binding to FMR1 gene was analyzed and quantified. Previous studies have approached this issue through indirect methods, ie, with a plasmid construct carrying the CGG expansion.17 Taking a more direct approach, we demonstrated that CGGBP1 binds to FMR1 gene in WT, PM, FXS and UFM cells. Direct CGGBP1 binding was observed only to unmethylated CGGs, with premutated and UFM alleles showing more binding compared with WT alleles. In FXS cell lines the binding was practically absent. A cell line derived from an FXS mosaic, including a premutation and a fully methylated mutation (FXS3), was employed in order to discriminate how the methyl group could affect CGGBP1 binding. Actually, despite the presence of a premutated allele in this line, the results indicated that this cell line acts more like a full mutation than as a premutation. In fact there was no CGGBP1 binding to FMR1 in this cell line. Therefore, our results obtained by ChIP experiments confirmed previous findings, indicating that CpG methylation prevents CGGBP1 binding.21 We then tested whether pharmacological DNA demethylation by use of 5-azadC could restore CGGBP1 binding to FMR1 in FXS cells. Cells treated with 5-azadC showed rescue of CGGBP1 binding to the FMR1 regulatory region (promoter and exon 1). Previous experiments had demonstrated that methylation of H3K9 is unaffected by 5-azadC treatment, while the euchromatic marker H3K4 becomes methylated.23 So, regardless of the chromatin status, CGGBP1 is drawn to the CGG unmethylated repeats and is able to bind this sequence. This finding is less obvious than it would appear. As described in more detail in a previous study,27 we observed that another protein, CTCF protein, does not bind to methylated CGGs and that its binding is not restored after pharmacological demethylation of FMR1 gene with 5-azadC in FXS cell lines.
The subsequent step was to investigate the effects of CGGBP1 transcript silencing. Knockdown experiments demonstrated that the absence of CGGBP1 mRNA apparently does not affect FMR1 transcription. At variance with previous observations,21 none of our cell lines, under the experimental condition, showed a decrease of FMR1 transcript in absence of CGGBP1 mRNA. The CGGBP1 depletion was achieved in two different silencing settings (transient and stable) and with two different methods (chemical transfections of esiRNA and lentiviral infections of shRNA) and was confirmed by western blot analysis (the CGGBP1 protein was practically undetectable, while FMRP levels were unchanged).
In conclusion, our results suggest that CGGBP1 is unlikely to be a direct transcriptional regulator of FMR1 transcription. However, CGGBP1 is strongly linked to the CGG tract of the FMR1 gene, when this is unmethylated or de novo demethylated, suggesting its possible implication in FMR1 regulation. Actually our observations lead to a different question: is it possible that CGGBP1 is involved in the stability of FMR1 CGG triplets? One of our attempts was in fact to establish stable CGGBP1-deficient clones that could be used to investigate the secondary effects on FMR1 gene expansion stability. The few clones obtained from stable transfection of FXS and UFM demonstrated that CGGBP1 depletion did not affect CGG expansion stability in expanded alleles. Further experiments are needed to validate this latter result and to investigate the meaning of CGGBP1 binding to FMR1.
References
Pirozzi F, Tabolacci E, Neri G : The FRAXopathies: definition, overview, and update. Am J Med Genet 2011; 155A: 1803–1816.
Bassell GJ, Warren ST : Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 2008; 60: 201–214.
Coffee B, Keith K, Albizua I et al: Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am J Hum Genet 2009; 85: 503–514.
Crawford DC, Meadows KL, Newman JL et al: Prevalence and phenotype consequence of FRAXA and FRAXE alleles in a large, ethnically diverse, special education-needs population. Am J Hum Genet 1999; 64: 495–507.
Sutcliffe JS, Nelson DL, Zhang F et al: DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet 1992; 1: 397–400.
Pietrobono R, Tabolacci E, Zalfa F et al: Molecular dissection of the events leading to inactivation of the FMR1 gene. Hum Mol Genet 2005; 14: 267–277.
Tabolacci E, Moscato U, Zalfa F et al: Epigenetic analysis reveals a euchromatic configuration in the FMR1 unmethylated full mutations. Eur J Hum Genet 2008; 16: 1487–1498.
Darnell JC, Van Driesche SJ, Zhang C et al: FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2001; 146: 247–261.
Smeets HJ, Smits AP, Verheij CE et al: Normal phenotype in two brothers with a full FMR1 mutation. Hum Mol Genet 1995; 4: 2103–2108.
Eiges R, Urbach A, Malcov M et al: Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell 2007; 1: 568–577.
Willemsen R, Bontekoe CJ, Severijnen LA et al: Timing of the absence of FMR1 expression in full mutation chorionic villi. Hum Genet 2002; 110: 601–605.
Kumari D, Usdin K : The distribution of repressive histone modifications on silenced FMR1 alleles provides clues to the mechanism of gene silencing in fragile X syndrome. Hum Mol Genet 2010; 19: 4634–4642.
Nolin SL, Sah S, Glicksman A et al: Fragile X AGG analysis provides new risk predictions for 45-69 repeat alleles. Am J Med Genet 2013; 161A: 771–778.
Volle CB, Delaney S : AGG/CCT interruptions affect nucleosome formation and positioning of healthy-length CGG/CCG triplet repeats. BMC Biochem 2013; 14: 33.
Hagihara M, He H, Kimura M et al: A small molecule regulates hairpin structures in d(CGG) trinucleotide repeats. Bioorg Med Chem Lett 2012; 22: 2000–2003.
Deissler H, Wilm M, Genç B et al: Rapid protein sequencing by tandem mass spectrometry and cDNA cloning of p20-CGGBP. A novel protein that binds to the unstable triplet repeat 5′-d(CGG)n-3′ in the human FMR1 gene. J Biol Chem 1997; 272: 16761–16768.
Naumann F, Remus R, Schmitz B et al: Gene structure and expression of the 5′-(CGG)(n)-3′-binding protein (CGGBP1). Genomics 2004; 83: 106–118.
Singh U, Westermark B : CGGBP1 is a nuclear and midbody protein regulating abscission. Exp Cell Res 2011; 317: 143–150.
Singh U, Roswall P, Uhrbom L et al: CGGBP1 regulates cell cycle in cancer cells. BMC Mol Biol 2011; 12: 28.
Singh U, Maturi V, Jones RE et al: CGGBP1 phosphorylation constitutes a telomere-protection signal. Cell Cycle 2014; 13: 96–105.
Müller-Hartmann H, Deissler H, Naumann F et al: The human 20-kDa 5′-(CGG)(n)-3′-binding protein is targeted to the nucleus and affects the activity of the FMR1 promoter. J Biol Chem 2000; 275: 6447–6452.
Chiurazzi P, Pomponi MG, Willemsen R et al: In vitro reactivation of the FMR1 gene involved in fragile X syndrome. Hum Mol Genet 1998; 7: 109–113.
Tabolacci E, Pietrobono R, Moscato U et al: Differential epigenetic modifications in the FMR1 gene of the fragile X syndrome after reactivating pharmacological treatments. Eur J Hum Genet 2005; 13: 641–648.
Pietrobono R, Pomponi MG, Tabolacci E et al: Quantitative analysis of DNA demethylation and transcriptional reactivation of the FMR1 gene in fragile X cells treated with 5-azadeoxycytidine. Nucleic Acids Res 2002; 30: 3278–3285.
Sinden RR : Biological implications of the DNA structures associated with disease-causing triplet repeats. Am J Hum Genet 1999; 64: 346–353.
Colak D, Zaninovic N, Cohen MS et al: Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 2014; 343: 1002–1005.
Lanni S, Goracci M, Borrelli L et al: Role of CTCF protein in regulating FMR1 locus transcription. PLoS Genet 2013; 9: e1003601.
Acknowledgements
This work was supported by Telethon grant (GGP10150), FRAXA Foundation and Italian Association for Fragile X syndrome to GN. We gratefully acknowledge Prof. Maurizio Genuardi for his critical revision of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Goracci, M., Lanni, S., Mancano, G. et al. Defining the role of the CGGBP1 protein in FMR1 gene expression. Eur J Hum Genet 24, 697–703 (2016). https://doi.org/10.1038/ejhg.2015.182
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.182
