
Abstract
Next-generation sequencing (NGS) technologies are now routinely used for gene identification in Mendelian disorders. Setting up cost-efficient NGS projects and managing the large amount of variants remains, however, a challenging job. Here we provide insights in the decision-making processes before and after the use of NGS in gene identification studies. Genetic factors are thought to have a role in ~70% of all epilepsies, and a variety of inheritance patterns have been described for seizure-associated gene defects. We therefore chose epilepsy as disease model and selected 35 NGS studies that focused on patients with a Mendelian epilepsy disorder. The strategies used for gene identification and their respective outcomes were reviewed. High-throughput NGS strategies have led to the identification of several new epilepsy-causing genes, enlarging our knowledge on both known and novel pathomechanisms. NGS findings have furthermore extended the awareness of phenotypical and genetic heterogeneity. By discussing recent studies we illustrate: (I) the power of NGS for gene identification in Mendelian disorders, (II) the accelerating pace in which this field evolves, and (III) the considerations that have to be made when performing NGS studies. Nonetheless, the enormous rise in gene discovery over the last decade, many patients and families included in gene identification studies still remain without a molecular diagnosis; hence, further genetic research is warranted. On the basis of successful NGS studies in epilepsy, we discuss general approaches to guide human geneticists and clinicians in setting up cost-efficient gene identification NGS studies.
Similar content being viewed by others

Introduction
Unraveling the genetic cause of a patient’s suspected inherited disorder is one of the major practices in human genetic research. Not only will the genetic background and cellular pathways involved in a specific disorder contribute to our understanding of human physiology, it will also influence the accuracy of prognosis, guidance in treatment decisions, and genetic counseling.
As Mendelian or monogenic inherited disorders are caused by a single gene defect, they form a unique model for assessing direct cause-effect relationships. Traditional genetic approaches such as linkage analysis and candidate gene screenings have implicated causal genes in the etiology of many Mendelian inherited disorders (http://omim.org/). Since 2005, the availability of next-generation sequencing (NGS) technologies has revolutionized the field of human genetics. They have enabled high-throughput gene identification studies and introduced novel strategies to unravel the genetic etiology of Mendelian inherited disorders.1, 2, 3, 4
Since targeted resequencing of selected genes, whole-exome and -genome sequencing (WES/WGS) have become affordable, the number of individuals that are being studied has rapidly enlarged. Henceforth, more and more researchers have been generating, analyzing, and interpreting NGS data. Setting up NGS projects and managing the large amount of variants remains, however, a challenging job. By referring to recent studies that used NGS technologies to identify causal gene defects in individuals with a presumed Mendelian epilepsy disorder, we provide insights in how the field is evolving. By discussing several of the pitfalls, we also aim to illustrate how more successful and efficient NGS projects can be conducted.
Method
In ∼70% of epilepsy patients genetic factors are thought to underlie the seizure phenotype.5 Mendelian epilepsy syndromes are rare, but causal gene defects following all monogenic inheritance patterns have been described in a subset of patients. Moreover, complicating factors such as a reduced penetrance, phenocopies, and genetic heterogeneity are common (eg, refs 6, 7). Epilepsy is thus an ideal disease model to discuss the implications of NGS on gene identification in Mendelian disorders. With a Medline search using the key words epilepsy and, respectively, whole-genome, -exome, or -panel sequencing about 100 reports were obtained. Thirty-five were selected based on the NGS methodology used to identify gene defects associated with the patient(s) epilepsy phenotype (ie, study design). The overall study design for gene identification in Mendelian disorders is commonly determined by the characteristics of the investigated phenotype (such as severity, onset age, and incidence) in combination with the structure of the pedigree (including consanguinity, gender, and number of affected individuals per generation). Together, this information is suggestive for the most likely mode of inheritance, which in turn points towards a starting NGS strategy. The most frequently used strategies are further referred to as the autosomal recessive (AR) homozygous, AR compound heterozygous, autosomal dominant (AD) heterozygous, AD de novo, and candidate approach strategy.1 Despite the importance of a good starting strategy, NGS permits to investigate different hypotheses in a single experiment.
NGS findings in Mendelian epilepsy disorders
All reported studies have been successful in associating a single gene defect with a Mendelian epilepsy disorder. Table 1 gives an overview of the selected 35 studies, their methodology, starting strategy, and findings. A study was considered ‘successful in gene identification’ in the following situations: (1) a variant was found in a gene previously associated with the same phenotype as that under investigation (ie, known gene−known phenotype); (2) a variant was found in a gene previously associated with another neuronal phenotype as that under investigation (ie, known gene−novel phenotype); (3) a variant was found in a gene not previously associated with any phenotype, but conclusive genetic evidence and/or substantial functional evidence was provided to support a causal relation with the phenotype (ie, novel gene−novel phenotype). Variants found in a single individual without any further evidence of causality are considered ‘candidate genes’.
Family based studies in monogenic epilepsy disorders searching for either AR or AD variants seemingly lead to a molecular diagnosis for nearly 100% of the included patients. Naturally this is just apparent, as negative reports are rarely published for family studies. Of note, when NGS identifies a known gene−known phenotype variant missed by traditional genetic approaches, the family will also not be reported. This publication bias makes it difficult to estimate the true success rate in family based studies; so what can we expect? On the basis of our experience, we are able to identify a (likely) pathogenic variant in ~25% of AD families and up to 40% of AR families (unpublished data). Large-scale studies on isolated patients or those who only include the index case in the initial analysis often do report positive and negative results simultaneously. This leads to a more accurate estimation of the success rate, which ranges from 10 to 48%.8, 9, 10, 11 Most of the early reports are, however, enriched for patient samples prescreened for established epilepsy genes, lowering the calculated success rate. Similar to our own experiences, the more recent NGS studies – including patients without preceding molecular investigations − provide a molecular diagnosis for ~30% of the investigated epilepsy patients.12, 13
Setting up NGS gene identification studies
The power of NGS in gene identification studies is clearly illustrated by the progress in epilepsy genetics. NGS can partially overcome the large clinical heterogeneity, genetic variability, and other complicating factors often seen in Mendelian disorders. The current results should nevertheless caution researchers and clinicians. They imply that up to 70% of the patients remain without a molecular diagnosis after inclusion in a NGS-based gene identification study. High-throughput is not an equivalent for infallibility, therefore we will discuss some of the considerations that have to be made when conducting a gene identification study using NGS.
Sampling and phenotyping
When setting up any genetic study, sample collection is the first and most important step: DNA material of key family members will be a necessity for interpretation of candidate variants. Although this might seem obvious, it is often not evident. Cooperation of both affected and unaffected family members is warranted, along with high quality and quantity of the DNA samples. A huge number of families are estranged from each other, either by distance or disputes. In addition, needle phobia is quite common but can be overcome by extracting DNA from saliva; a technique that also allows sampling of very young children. Manual extraction and purification of a sufficient amount of saliva can render the required DNA material necessary for NGS.14
Collaborating with motivated clinicians is therefore key for starting medical genetic research. Also, efficient gene identification goes hand in hand with deep phenotyping. A specific gene defect can be associated with different types of neurological phenotypes. Vice versa, different gene defects can be associated with the same phenotype. These obstacles can partially be overcome by NGS and might support the idea ‘to sequencing everyone’. Specific clinical hallmarks can nevertheless be indicative for a defined genetic syndrome and subsequent genetic defect. Selecting phenotypically matched cases can thus enlarge the chance of finding independent variants in the same gene (eg, ref. 15).
Overall, the rise of NGS technologies has broadened the focus of study populations. Large multi-generation families were favored in the past, in which a combined approach of linkage analysis with NGS of positional candidate genes has become the major approach. Problems including reduced penetrance and variable expression within these families remain, however, problematic. Identifying highly penetrant variants in patients with a severe phenotype, either presenting as sporadic patients (ie, de novo analyses) or as siblings (ie, de novo analyses with germline mosaicism or recessive inheritance), is more straightforward. Hereby making isolated cases and sib pairs most popular in NGS studies.
NGS strategy
After collecting and thoroughly assessing the study cohort, the most likely inheritance pattern can be determined for each family. Figure 1 is provided as a guide to help deciding which strategy can best be applied to analyze specific sample sets. The choice of strategy will define which individuals should be sequenced: a trio-approach is required for a straightforward de novo analysis, whereas studies on families with multiple affected individuals are less indicative towards the subjects to be sequenced. The earliest NGS studies showed that including sequencing data of an increasing number of family members gives considerably more power to any genomic analysis.16 Depending on the pedigree structure some individuals will, however, be more informative than others. Several tools exist for automated selection, such as the statistical framework GIGI-Pick, although they mainly focus on large and complex pedigrees.17 Studies including AD families mostly opt for sequencing two or three distantly related relatives and look to identify variants according to the kinship. Alternatively, some studies select unrelated but phenotypically or genetically (through linkage analysis) matched index cases, and search for a shared gene defect. As gene identification in AR families relies on the presence of two hits in the same gene, sequencing one or two individuals is often sufficient in order to reach a molecular diagnosis.

Visual aid to the guide NGS projects. Orange boxes are indicators for the project strategy. Blue boxes indicate to choose a sequencing technique; depending on this choice different results will be obtained. Green boxes contain the different project strategies. Red lines indicate the minimum sample requirement, sequencing more individuals of the same family will render more power, but is not necessary to find a molecular diagnosis in the majority of patients.
Conclusive genetic proof of pathogenicity can only be established when the same gene defect is identified in independent patients. The yield of pathogenic variants in established epilepsy genes and for specific patient subgroups can, however, vary from <1% to up to 80%. Phenotypic and genetic heterogeneity further complicate an accurate estimation of the required amount of patients to sequence, in order to find variants in the same gene. Data sharing between genetic centers is indispensable in the future to achieve a higher yield of molecular diagnoses (eg, refs 11, 18). Several initiatives have recently been set-up to facilitate multi-center collaborations between genetic diagnostic and research center (eg, ref. 19, https://genematcher.org/, and http://www.matchmakerexchange.org/).
Sequencing
The 35 selected studies have collectively sequenced 1895 individuals belonging to 1244 different epilepsy families on a NGS platform. WES is by far the most used sequencing technique in research: 73% compared with 11% WGS and 16% targeted resequencing. As the exome represents an enriched part of the genome for disease-associated variants, this preference is justifiable in gene identification studies. Loss of information will nevertheless have to be taken into account, because not the complete exome will be optimally covered. Owing to dropping sequencing costs and optimization of bioinformatics tools, WGS is most likely to take over in the future.20 In the mean while, targeted resequencing can also provide a higher coverage of selected regions. For large families where linkage data are available, the combination with targeted resequencing can be very powerful. Remarkably, WES is also preferred when linkage data are available (Table 1: 5/8 studies). The time needed to optimize custom gene panels, in combination with having direct access to the entire exome if no (likely) pathogenic variant can be found in the captured region(s) probably underlies this choice. The increasing speed and decreasing cost have also led to the implementation of NGS in clinical genetic services. Targeted gene panels long had an ethical and cost-efficient advantage over WES/WGS by screening only the relevant disease genes.21, 22, 23 Owing to their limited power in variant yield for heterogeneous disorders (such as epilepsy), diagnostic labs are now moving towards the use of WES/WGS.24, 25 Validation of candidate variants and segregation analysis in additional family members is generally still done by direct Sanger sequencing.
Nowadays, NGS data is often generated in service facilities, therefore the actual sequencing procedure will not be further discussed here. Differences in variant callings due to capturing, sequencing technology, and mapping algorithms have been extensively reported.26, 27, 28 To handle and analyze the generated data most labs have also developed custom pipelines over the years. If such pipeline is not available, we recommend the user-friendly and web-based tool Galaxy and Broad’s Genome Analysis Toolkit (GATK) variant caller to compile, annotate, and analyze data.
NGS variant management
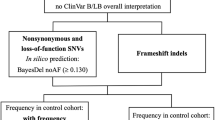
After collecting the right samples, determining whom to sequence and generating the data, most of the work still has to start. Interpretation of NGS data and selecting the causal gene defect has become more time consuming than the data generation itself. A set of highly recommended guidelines for interpretation of sequence variants was recently published by the ACMG. They include comprehensive lists of (I) population, disease, and gene specific databases, (II) in silico prediction algorithms, and (III) criteria for classifying candidate variants.29 Here we further discuss some general principles of a standard filtering cascade used during variant prioritization (Figure 2).

General filtering cascade. NGS data sets generate a large amount of variants. A standard prioritization scheme consists of four different layers, which are further separated into variant and gene based: (1) a variant selection is based on quality, frequency in a control population and predicted impact on the protein; (2) based on the assumed mode of inheritance the genetics selection can reduce the number of candidate variants; (3) extensive literature study on the genes harboring, the remaining variants can lead to a reduction of candidate genes; (4) different tools can be used to help interpret the possible impact of the gene defects and assess the most likely associations with the phenotype of interest.
A variant-based selection on the quality of the variant calling (eg, coverage), frequency in population databases and predicted impact on the encoded protein is usually the first step. Setting quality cutoffs pursues an optimal balance between sensitivity and specificity. Trying different settings and analyzing multiple samples side-by-side will prove useful to reduce false-positive and false-negative results. In addition, joint calling of different samples versus single sample calling will generally provide better quality calls. The most widely used variant caller, GATK, has an active community that is constantly guiding users in how to optimize quality thresholds. We encourage new users to participate actively (http://gatkforums.broadinstitute.org/). Cutoffs regarding the frequency and predicted impact of a variant will mainly depend on the assumed inheritance pattern and incidence of the investigated phenotype. For example, when looking for de novo variants in an individual with a devastating phenotype, candidate variants should be absent in control databases. On the contrary, when looking for AR or AD variants with a reduced penetrance a certain percentage of heterozygous carriers can be expected. Publicly available databases including the most recently released ExAC browser (http://exac.broadinstitute.org, Cambridge, MA, USA), are constantly expanding. Yet, setting up an in-house variant database is equally important as it has the additional advantage to enable exclusion of NGS platform-dependent variants. It can furthermore provide information on variant frequencies in distinct ethnical groups.
A second variant-based prioritization, here referred to as genetic-selection, is based on the assumptions made regarding inheritance patterns. Although assumptions regarding the mode of inheritance is the most empirical method to identify disease-causing variants, it can be misleading. Analysis of the data under different inheritance models is recommended, as has been nicely illustrate in several recent NGS studies (eg, refs 3, 13).
Next, data mining on all genes harboring remaining candidate variants will enable researchers to prioritize genes of interest. Several programs can help to rapidly pinpoint the genes most likely associated with the investigated phenotype (eg, ToppGene and Biograph) or evaluate specific expression patterns (eg, EvoTol, GTex, and Human Protein Atlas). Of note, data mining is quite subjective and often correlates with the experience of the researcher. Automated probability interpretations have the advantage of speed and throughput over manual labor, but share a large bias towards existing knowledge on gene function and disease pathomechanisms. Therefore it is recommended to level different programs and manually check the ranked candidate genes.
The final interpretation about the potential impact of a specific gene defect is partly variant based and partly gene based. Ideally such interpretation is supported by in vitro or in vivo experiments, but owing to practical considerations in silico predictions can provide a first and fast indication. The recently released integrated framework combined annotation-dependent depletion unites the most widely used interpretation tools. Again a bias towards current knowledge and the limitation of most in silico tools towards variants with a potentially large effect (eg, protein-altering variants) has to be taken into account. Several scoring systems also exists for ranking genes according to their tolerance for genetic variations or likelihood to be involved in disease pathways. These work quite well for de novo variants, but are less indicative when examining recessive gene defects. The existence of multiple transcripts and variable expression patterns can furthermore cause contradictions between predictions algorithms.
It is clear that NGS variant management is not a rigid science for which a standard protocol can be followed. General methodologies for prioritizing and interpreting variants are nevertheless proposed. Depending on the nature of the gene identification study, different cutoffs will proof most valuable. Getting acquainted with frequently used annotation sets and prediction tools is necessary when learning how to interpret NGS data. Basic genetic ideas remain valid in NGS studies and researchers should always be critical about early assumptions: there are always several options and NGS gives you the power to investigate them all trough a single experiment. Finally, an interpretation will have to be made for determining the nature of a variant based on combined genetic, clinical, and preferably functional data.30
Technical pitfalls
Even when multiple individuals of the same family are sequenced, the data is analyzed under different inheritance models, and filtering strategies have been worn out, it remains possible that no variant is interpreted as (likely) pathogenic. When working with NGS technologies it is important to realize that no single technique will cover everything. Identification of somatic and germline mosaic events or copy number variants remain complicated despite the progress made in the field.31, 32 Copy number variants have an important contribution to many Mendelian disorders33, 34, 35 and are often overlooked or disregarded in NGS studies. The pathogenic variant can also be unsequenced due to lack of targeting, capturing, or bad mapping quality. Variant callers and annotation tools might furthermore be wrong or misleading. A very useful tool to check the nomenclature of a variant, together with its impact, is the Mutalyzer 2.0.9.36 Ideally, each individual should be sequenced and analyzed on multiple platforms to reduce the chance of missing causal variants due to technical errors.28 Unfortunately this is not a realistic situation; most researchers have a budget that is preferably used to investigate as many families as possible. Analyzing NGS data with different variant callers and annotation tools is for the moment the best way to partially compensate for limited resources.
Conclusion
High-throughput screenings have led to the identification of many new disease-causing genes for Mendelian diseases. The large phenotypical and genetic heterogeneity of human disorders will nevertheless make it necessary to collaborate on a large scale and pool data in a joined effort to support causality of specific gene defects (eg, ref. 11). In the end, accumulating genetic information will expand our knowledge on normal human body functions and eventually lead to a better understanding of disease pathways.
NGS is a continuous evolving field that is becoming increasingly available, faster and cheaper. Henceforth, more and more researchers will be setting up NGS projects to give a molecular diagnosis to their patient(s). By summarizing recent findings in patients with Mendelian epilepsy disorders and discussing the used NGS research designs, we highlight several considerations that have to be taken into account before and during a gene identification study. It is clear that NGS has provided many insights and opportunities in genetic research, but with still more than half of the patients without a molecular diagnosis, further genetic research is a priority.
Ethical guidelines
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
Gilissen C, Hoischen A, Brunner HG et al: Disease gene identification strategies for exome sequencing. Eur J Hum Genet 2012; 20: 490–497.
Rabbani B, Mahdieh N, Hosomichi K et al: Next-generation sequencing: impact of exome sequencing in characterizing Mendelian disorders. J Hum Genet 2012; 57: 621–632.
Yang Y, Muzny DM, Xia F et al: Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 2014; 312: 1870–1879.
Koboldt DC, Steinberg KM, Larson DE et al: The next-generation sequencing revolution and its impact on genomics. Cell 2013; 155: 27–38.
Hildebrand MS, Dahl HH, Damiano JA et al: Recent advances in the molecular genetics of epilepsy. J Med Genet 2013; 50: 271–279.
Escayg A, MacDonald BT, Meisler MH et al: Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet 2000; 24: 343–345.
Claes S, Devriendt K, Lagae L et al: The X-linked infantile spasms syndrome (MIM 308350) maps to Xp11.4-Xpter in two pedigrees. Ann Neurol 1997; 42: 360–364.
Allen AS, Berkovic SF, Cossette P et al: De novo mutations in epileptic encephalopathies. Nature 2013; 501: 217–221.
Michaud JL, Lachance M, Hamdan FF et al: The genetic landscape of infantile spasms. Hum Mol Genet 2014; 23: 4846–4858.
Lemke JR, Riesch E, Scheurenbrand T et al: Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia 2012; 53: 1387–1398.
EuroEPINOMICS-RES Consortium, Epilepsy Phenome/Genome Project, Epi4K Consortium: De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet 2014; 95: 360–370.
Mercimek-Mahmutoglu S, Patel J, Cordeiro D et al: Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia 2015; 56: 707–716.
Muona M, Berkovic SF, Dibbens LM et al: A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat Genet 2015; 47: 39–46.
Kidd JM, Sharpton TJ, Bobo D et al: Exome capture from saliva produces high quality genomic and metagenomic data. BMC Genomics 2014; 15: 262–278.
Suls A, Jaehn JA, Kecskes A et al: De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am J Hum Genet 2013; 93: 967–975.
Roach JC, Glusman G, Smit AF et al: Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science 2010; 328: 636–639.
Cheung CY, Marchani BE, Wijsman EM : A statistical framework to guide sequencing choices in pedigrees. Am J Hum Genet 2014; 94: 257–267.
Zhang X, Ling J, Barcia G et al: Mutations in QARS, encoding glutaminyl-tRNA synthetase, cause progressive microcephaly, cerebral-cerebellar atrophy, and intractable seizures. Am J Hum Genet 2014; 94: 547–558.
Fokkema IF, Taschner PE, Schaafsma GC et al: LOVD v.2.0: the next generation in gene variant databases. Hum Mutat 2011; 32: 557–563.
Gilissen C, Hehir-Kwa JY, Thung DT et al: Genome sequencing identifies major causes of severe intellectual disability. Nature 2014; 511: 344–347.
Sun Y, Ruivenkamp CA, Hoffer MJ et al: Next-generation diagnostics: gene panel, exome, or whole genome? Hum Mutat 2015; 36: 648–655.
Vrijenhoek T, Kraaijeveld K, Elferink M et al: Next-generation sequencing-based genome diagnostics across clinical genetics centers: implementation choices and their effects. Eur J Hum Genet 2015; 23: 1142–1150.
Lohmann K, Klein C : Next generation sequencing and the future of genetic diagnosis. Neurotherapeutics 2014; 11: 699–707.
Della ME, Ciccone R, Brustia F et al: Improving molecular diagnosis in epilepsy by a dedicated high-throughput sequencing platform. Eur J Hum Genet 2014; 23: 354–362.
Wang J, Gotway G, Pascual JM et al: Diagnostic yield of clinical next-generation sequencing panels for epilepsy. JAMA Neurol 2014; 71: 650–651.
Metzker ML : Sequencing technologies - the next generation. Nat Rev Genet 2010; 11: 31–46.
Clark MJ, Chen R, Lam HY et al: Performance comparison of exome DNA sequencing technologies. Nat Biotechnol 2011; 29: 908–914.
O'Rawe J, Jiang T, Sun G et al: Low concordance of multiple variant-calling pipelines: practical implications for exome and genome sequencing. Genome Med 2013; 5: 28.
Richards S, Aziz N, Bale S et al: Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–423.
Sunyaev SR : Inferring causality and functional significance of human coding DNA variants. Hum Mol Genet 2012; 21: R10–R17.
Rios JJ, Delgado MR : Using whole-exome sequencing to identify variants inherited from mosaic parents. Eur J Hum Genet 2014; 23: 547–550.
Tan R, Wang Y, Kleinstein SE et al: An evaluation of copy number variation detection tools from whole-exome sequencing data. Hum Mutat 2014; 35: 899–907.
Mefford HC, Yendle SC, Hsu C et al: Rare copy number variants are an important cause of epileptic encephalopathies. Ann Neurol 2011; 70: 974–985.
Steffens M, Leu C, Ruppert AK et al: Genome-wide association analysis of genetic generalized epilepsies implicates susceptibility loci at 1q43, 2p16.1, 2q22.3 and 17q21.32. Hum Mol Genet 2012; 21: 5359–5372.
de Kovel CG, Trucks H, Helbig I et al: Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain 2010; 133: 23–32.
Wildeman M, van OE, den Dunnen JT et al: Improving sequence variant descriptions in mutation databases and literature using the mutalyzer sequence variation nomenclature checker. Hum Mutat 2008; 29: 6–13.
Vaher U, Noukas M, Nikopensius T et al: De novo SCN8A mutation identified by whole-exome sequencing in a boy with neonatal epileptic encephalopathy, multiple congenital anomalies, and movement disorders. J Child Neurol 2013; 29: NP202–NP206.
Veeramah KR, Johnstone L, Karafet TM et al: Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia 2013; 54: 1270–1281.
Hackenberg A, Baumer A, Sticht H et al: Infantile epileptic encephalopathy, transient choreoathetotic movements, and hypersomnia due to a de novo missense mutation in the SCN2A gene. Neuropediatrics 2014; 45: 261–264.
Nakajima J, Okamoto N, Tohyama J et al: De novo EEF1A2 mutations in patients with characteristic facial features, intellectual disability, autistic behaviors and epilepsy. Clin Genet 2014; 87: 356–361.
Vanderver A, Simons C, Schmidt JL et al: Identification of a novel de novo p.Phe932Ile KCNT1 mutation in a patient with leukoencephalopathy and severe epilepsy. Pediatr Neurol 2014; 50: 112–114.
Baasch AL, Huning I, Gilissen C et al: Exome sequencing identifies a de novo SCN2A mutation in a patient with intractable seizures, severe intellectual disability, optic atrophy, muscular hypotonia, and brain abnormalities. Epilepsia 2014; 55: e25–e29.
Lee H, Lin MC, Kornblum HI et al: Exome sequencing identifies de novo gain of function missense mutation in KCND2 in identical twins with autism and seizures that slows potassium channel inactivation. Hum Mol Genet 2014; 23: 3481–3489.
Saitsu H, Kato M, Osaka H et al: CASK aberrations in male patients with Ohtahara syndrome and cerebellar hypoplasia. Epilepsia 2012; 53: 1441–1449.
Nakamura K, Kodera H, Akita T et al: De Novo mutations in GNAO1, encoding a Galphao subunit of heterotrimeric G proteins, cause epileptic encephalopathy. Am J Hum Genet 2013; 93: 496–505.
Barcia G, Fleming MR, Deligniere A et al: De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet 2012; 44: 1255–1259.
Milh M, Falace A, Villeneuve N et al: Novel compound heterozygous mutations in TBC1D24 cause familial malignant migrating partial seizures of infancy. Hum Mutat 2013; 34: 869–872.
Nakamura K, Osaka H, Murakami Y et al: PIGO mutations in intractable epilepsy and severe developmental delay with mild elevation of alkaline phosphatase levels. Epilepsia 2014; 55: e13–e17.
Paciorkowski AR, Weisenberg J, Kelley JB et al: Autosomal recessive mutations in nuclear transport factor KPNA7 are associated with infantile spasms and cerebellar malformation. Eur J Hum Genet 2013; 22: 587–593.
Hitomi Y, Heinzen EL, Donatello S et al: Mutations in TNK2 in severe autosomal recessive infantile onset epilepsy. Ann Neurol 2013; 74: 496–501.
Campeau PM, Kasperaviciute D, Lu JT et al: The genetic basis of DOORS syndrome: an exome-sequencing study. Lancet Neurol 2014; 13: 44–58.
Basel-Vanagaite L, Hershkovitz T, Heyman E et al: Biallelic SZT2 mutations cause infantile encephalopathy with epilepsy and dysmorphic corpus callosum. Am J Hum Genet 2013; 93: 524–529.
Poduri A, Heinzen EL, Chitsazzadeh V et al: SLC25A22 is a novel gene for migrating partial seizures in infancy. Ann Neurol 2013; 74: 873–882.
Pippucci T, Parmeggiani A, Palombo F et al: A novel null homozygous mutation confirms CACNA2D2 as a gene mutated in epileptic encephalopathy. PLoS One 2013; 8: e82154.
Heron SE, Smith KR, Bahlo M et al: Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet 2012; 44: 1188–1190.
Ishida S, Picard F, Rudolf G et al: Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat Genet 2013; 45: 552–555.
Dibbens LM, de Vries B, Donatello S et al: Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet 2013; 45: 546–551.
Edvardson S, Oz S, Abulhijaa FA et al: Early infantile epileptic encephalopathy associated with a high voltage gated calcium channelopathy. J Med Genet 2013; 50: 118–123.
Alazami AM, Hijazi H, Kentab AY et al: NECAP1 loss of function leads to a severe infantile epileptic encephalopathy. J Med Genet 2014; 51: 224–228.
Corbett MA, Bahlo M, Jolly L et al: A focal epilepsy and intellectual disability syndrome is due to a mutation in TBC1D24. Am J Hum Genet 2010; 87: 371–375.
De Fusco M, Vago R, Striano P et al: The alpha2B-adrenergic receptor is mutant in cortical myoclonus and epilepsy. Ann Neurol 2014; 75: 77–87.
Heron SE, Grinton BE, Kivity S et al: PRRT2 mutations cause benign familial infantile epilepsy and infantile convulsions with choreoathetosis syndrome. Am J Hum Genet 2012; 90: 152–160.
Carvill GL, Heavin SB, Yendle SC et al: Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet 2013; 45: 825–830.
Lemke JR, Hendrickx R, Geider K et al: GRIN2B mutations in west syndrome and intellectual disability with focal epilepsy. Ann Neurol 2013; 75: 147–154.
Lemke JR, Lal D, Reinthaler EM et al: Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat Genet 2013; 45: 1067–1072.
Martin HC, Kim GE, Pagnamenta AT et al: Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis. Hum Mol Genet 2014; 23: 3200–3211.
Touma M, Joshi M, Connolly MC et al: Whole genome sequencing identifies SCN2A mutation in monozygotic twins with Ohtahara syndrome and unique neuropathologic findings. Epilepsia 2013; 54: e81–e85.
Veeramah KR, O'Brien JE, Meisler MH et al: De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet 2012; 90: 502–510.
Lee HY, Huang Y, Bruneau N et al: Mutations in the novel protein PRRT2 cause paroxysmal kinesigenic dyskinesia with infantile convulsions. Cell Rep 2012; 1: 2–12.
Acknowledgements
To gain experience in NGS data analysis and gene identification in Mendelian epilepsies the involved researchers had the chance to be part of a fruitful, large-scale consortium. Therefore we thank the Eurocores program EuroEPINOMICS of the European Science Foundation (G.A.136.11.N and FWO/ESF-ECRP), all partners and in particular Rudi Balling and Patrick May of the Family Genomics group at the Institute for Systems Biology (Luxembourg). We also acknowledge the constant support of the Fund for Scientific Research Flanders (FWO), the University of Antwerp and the centralized services of the VIB-Department Molecular genetics with special mentioning of Peter De Rijk for developing our in-house NGS pipeline (GenomeComb; http://genomecomb.sourceforge.net/). KH is a PhD fellow of the Institute for Science and Technology (IWT)-Flanders and AS is a postdoctoral fellow of the FWO.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Hardies, K., Weckhuysen, S., De Jonghe, P. et al. Lessons learned from gene identification studies in Mendelian epilepsy disorders. Eur J Hum Genet 24, 961–967 (2016). https://doi.org/10.1038/ejhg.2015.251
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.251
This article is cited by
-
The role of excitatory amino acid transporter 2 (EAAT2) in epilepsy and other neurological disorders
Metabolic Brain Disease (2023)
-
Molecular cloning and characterization of the family of feline leucine-rich glioma-inactivated (LGI) genes, and mutational analysis in familial spontaneous epileptic cats
BMC Veterinary Research (2017)
-
Unexplained Early Infantile Epileptic Encephalopathy in Han Chinese Children: Next-Generation Sequencing and Phenotype Enriching
Scientific Reports (2017)
-
Lessons from genetics: is it time to revise the therapeutic approach to children with steroid-resistant nephrotic syndrome?
Journal of Nephrology (2016)
