
Abstract
LMNA encodes lamin A and lamin C, two major components of the nuclear lamina, and its pathogenic variants lead to a dozen distinct clinical entities collectively known as laminopathies. Most LMNA-related laminopathies are autosomal dominant but four are autosomal recessive; furthermore, some of the dominant variants have been associated with distinct phenotypes when inherited recessively, further complicating the ability to correlate genotype with phenotype. We report a consanguineous family in which the index presented with an apparently unique constellation of poikiloderma, joint motion restriction and distal acroosteolysis but lacks features of muscle weakness, lipodystrophy, or cardiac or craniofacial involvement. Molecular analysis revealed the presence of a novel homozygous LMNA missense variant (NM_170707.3:c.1774G>A; p.(Gly592Arg)) within an area of autozygome that is not shared by his unaffected siblings. The proposed causal link is further supported by in silico analysis of this variant. Our case suggests an expansion of LMNA allelic disorders to include distal acroosteolysis, poikiloderma and joint stiffness (DAPJ).
Similar content being viewed by others

Introduction
Lamins are highly conserved proteins that comprise the nuclear lamina, a dense proteinaceous meshwork beneath the inner nuclear membrane that was identified several decades ago.1 Lamins are classified as intermediate filament proteins and share the structural organization of an N-terminus head, a rod consisting of four alpha-helical domains with linkers interspersed between them, and a C-terminus tail.2 Although originally thought to serve a pure mechanical role supporting the three-dimensional structure of the nucleus, lamins are now known to have a wide range of physiological roles including chromatin organization, transcriptional regulation, and DNA damage repair and differentiation.3
Humans, like other mammals, have three lamin genes. LMNA encodes lamin A and lamin C by virtue of alternative splicing. Unlike LMNB1, which encodes lamin B1 and has been linked to a single human disease, LMNA ranks among human genes with the largest number of allelic disorders. LMNA-linked disorders (known as laminopathies) are mostly autosomal dominant but biallelic LMNA pathogenic variants are known to cause four diseases: restrictive dermopathy, Emery–Dreifuss muscular dystrophy (EDMD), mandibuloacral dysplasia and Charcot–Marie–Tooth disease, type 2B1.4, 5, 6, 7 In this report, we expand the number of LMNA-related laminopathies to include an apparently novel syndrome characterized by distal acroosteolysis, poikiloderma and joint stiffness.
Clinical report
We report a 9-year-old Saudi boy who was seen in rheumatology clinic after referral due to abnormal skin pigmentation and flexing contractures of his extremities. His parents are first degree cousins and have four healthy children (Figure 1). The patient is a product of full-term uneventful pregnancy. He was normal until the age of 3 years when he started to develop progressive hyperpigmented and hypopigmented skin macules around the large and small joints, buttocks, and on the face, especially around the eyes (Figure 1). Oral mucous membrane was spared and there was no associated skin erythema, pain or pruritus. The pigmentary skin changes gradually evolved to involve the dorsum of both hands and feet, with thinning of the skin of both hands and feet mainly the palms and the soles. He developed progressive restriction of movement over almost all joints caused by his dermopathy. The motion limitation of these joints was not accompanied by pain or swelling. There was a history of chronic constipation but with no associated anal fissures. His development (physical and cognitive) has always been normal and there was no history of muscle weakness, muscle aches, syncope or palpitation.

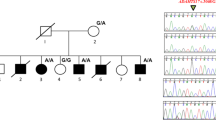
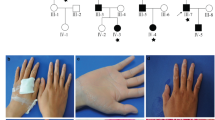
(a) Clinical and radiographic images of index. Note lack of craniofacial dysmorphism, the poikiloderma and its distribution as described in the text as well as the distal acroosteolysis. (b) Family pedigree. (c) Genome-wide autozygosity analysis shows that LMNA maps to an autozygous interval in the index not shared by the other family members (indicated by dark blue color). (d) Sequence chromatogram showing the LMNA mutation. (e) The Gly592 residue is conserved across species as shown in this multispecies alignment. (f) Diagrammatic representation of LMNA protein showing the location of the p.(Gly592Arg) variant in the C terminus.
On examination, he had normal growth parameters with weight of 32 kg (75th percentile), and height of 133.4 cm (75th percentile), and no craniofacial dysmorphism. Hypo- and hyperpigmented macules were noted over the periorbital region, metacarpophalangeal joints, elbows, axilla, knees, ankles and buttocks. He had scleroderma-like skin over the palms and the soles with old pressure ulcers over the soles. The very tight skin caused generalized joint stiffness with marked limitation of movement over elbows, wrists, ankles and small joints of the hands and feet. This caused impaired motor skills (such as difficulty in walking with waddling gait) and inability to perform fine movements of the hands (such as writing). The terminal phalanges of the hands were small and rounded but not swollen or tender (Figure 1). Hair was normal as was the fat distribution.
Laboratory investigations included CBC, LFT, ESR and muscle enzymes, which were all within normal limits. His fasting level of blood glucose was 4.3 (4.1–6.1 mmol/l), insulin 18.5 (2.6–24.9 uU/ml), cholesterol 3.9 (<5.2 mmol/l) and triglycerides 1.6 (<1.7 mmol/l). His serology evaluation was negative for ANA on multiple occasions, ENA, and Scleroderma 70Ab. Skeletal radiological survey revealed no mandibular or clavicular hypoplasia, diffuse osteopenia and resorption of the distal phalanges predominantly involving the hands, and to a lesser extent the feet. Nerve conduction study and EMG were normal as was MRI of the thigh muscles. Electrocardiogram and echocardiography were normal with no evidence of cardiomyopathy. Skin biopsy from a pigmentary skin lesion showed evidence of hyperkeratosis, basal layer hyperpigmentation, normal dermis but no evidence of inflammation. Clinical sequencing of ADAR1 for dyschromatosis symmetrica hereditaria (DSH), a condition that also exhibits a similar pattern of hyper/hypo-pigmentary macules, was negative.
Atypical juvenile dermatomyositis was initially suspected and he was treated with prednisolone, methotrexate and hydroxychloroquine but showed no response so they were discontinued. Currently the patient’s condition is static and he continues to have good growth. He is enrolled in a physical therapy program to relieve his joint stiffness and receives yearly cardiac evaluation.
Molecular studies
The patient, his siblings and parents were recruited using an IRB-approved research protocol with informed consent (KFSHRC RAC#2121053). He was tested using our recently published Mendeliome assay, a next-generation sequencing-based gene panel assay that comprises nearly all known OMIM disease genes.8 In addition, autozygome analysis for the entire family using genomewide SNP analysis and AutoSNPa was pursued as described before.9
Results
The Mendeliome assay revealed a homozygous change in LMNA as the only likely candidate variant. The change (NM_170707.3:c.1774G>A) predicts a novel nonsynonymous missense change (p.(Gly592Arg)) involving an invariant amino-acid residue in humans (ExAC and Exome Variant Server do not list any changes in Gly592). PolyPhen, SIFT and CADD assigned high pathogenic scores of 1, 0 and 33, respectively. As shown in Figure 1, this amino-acid residue is highly conserved among a number of species. Reassuringly, LMNA was within an autozygous interval that is exclusive to index (Figure 1). The variant fully segregated with the disease in an autosomal recessive manner in the family. It was also absent in 703 Saudi in-house whole exome database. We have submitted this variant to ClinVar: http://www.ncbi.nlm.nih.gov/clinvar/ (SCV accession number SCV000255936).
Discussion
LMNA-related laminopathies are surprisingly restricted in their pleiotropy to a small subset of tissues despite the ubiquitous distribution of lamin A and lamin C.10 Their predilection to skin, fat, bone, muscle and heart is one of the many unanswered questions about laminopathies and their pathogenesis. Another major conundrum in laminopathies is lack of genotype/phenotype correlation.11 One notorious exception is the ‘silent’ Hutchinson–Gilford progeria syndrome (HGPS) variant (NM_170707.3:c.1824C>T) that results in aberrant splicing and deletion of 50 amino acids near the C terminus. The resulting mutant protein retains the CAAX box but lacks the site for endoproteolytic cleavage. Several lines of evidence strongly support the view that this results in a toxic gain of function because the uncleaved protein is constitutively fernesylated, and this may explain the specific set of signs and symptoms that characterize HGPS.12 The genotype/phenotype correlation for most other laminopathy-related variants is very weak and multiple allelic laminopathies are known to co-exist within the same family.13, 14
The capacity of some LMNA variants to cause autosomal dominant versus recessive laminopathies is a phenomenon not yet completely understood. In fact, the same variants have been found to underlie dominant and recessive laminopathies in some instances, for example, NM_170707.3:c.1303C>T; p.(Arg435Cys) causes autosomal dominant dilated cardiomyopathy and autosomal recessive restrictive dermopathy.15, 16 Only four recessive laminopathies have been described to date: EDMD, restrictive dermopathy, mandibuloacral dysplasia and Charcot–Marie–Tooth disease, type2B1. The phenotype we describe in this report is clearly distinct from these disorders. The index has no muscle weakness, normal muscle enzymes, normal EMG and muscle MRI, normal EKG and echocardiogram; features that are highly inconsistent with EDMD. Similarly, the very few patients with LMNA-related restrictive dermopathy were born with typical features of profound developmental consequences of the abnormally inelastic skin tissue and the condition is uniformly fatal, hence the term ‘lethal restrictive dermopathy’.17 Although the index did have some features of mandibuloacral dysplasia, we are of the opinion that his phenotype is sufficiently different that labeling him as mandibuloacral dysplasia would be a gross distortion of his clinical diagnosis. Specifically, while joint stiffness, poikiloderma and distal acroosteolysis are known to occur in mandibuloacral dysplasia, our patient lacks many of the key features of the syndrome including: growth retardation, lipodystrophy, the typical bird-like facies with mandibular hypoplasia, hypoplasia of the clavicle, alopecia and hyperinsulinemia.18 Taken together, we propose DAPJ (distal acroosteolysis, poikiloderma and joint stiffness) as an acronym for the laminopathy we describe in this report to differentiate it from other laminopathies. Whether this phenotype is allele specific or can be caused by other LMNA variants can only be revealed by future reports.
References
Fawcett DW : On the occurrence of a fibrous lamina on the inner aspect of the nuclear envelope in certain cells of vertebrates. Am J Anat 1966; 119: 129–145.
Fisher DZ, Chaudhary N, Blobel G : cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc Natl Acad Sci 1986; 83: 6450–6454.
Gruenbaum Y, Foisner R : Lamins: nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu Rev Biochem 2015; 84: 131–164.
Schreiber KH, Kennedy BK : When lamins go bad: nuclear structure and disease. Cell 2013; 152: 1365–1375.
Worman HJ : Nuclear lamins and laminopathies. J Pathol 2012; 226: 316–325.
Worman HJ, Bonne G : “Laminopathies”: a wide spectrum of human diseases. Exp Cell Res 2007; 313: 2121–2133.
De Sandre-Giovannoli A, Chaouch M, Kozlov S et al: Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am J Hum Genet 2002; 70: 726–736.
Saudi Mendeliome Group: Comprehensive gene panels provide advantages over clinical exome sequencing for Mendelian diseases. Genome Biol 2015; 16: 134.
Alkuraya FS : Discovery of rare homozygous mutations from studies of consanguineous pedigrees. Curr Protoc Hum Genet 2012; 6: 12.
Lattanzi G, Benedetti S, Bertini E et al: Laminopathies: many diseases, one gene. Report of the first Italian Meeting Course on Laminopathies. Acta Myol 2011; 30: 138.
Carboni N, Politano L, Floris M et al: Overlapping syndromes in laminopathies: a meta-analysis of the reported literature. Acta Myol 2013; 32: 7.
Gordon LB, Rothman FG, López-Otín C, Misteli T : Progeria: a paradigm for translational medicine. Cell 2014; 156: 400–407.
Rankin J, Ellard S : The laminopathies: a clinical review. Clin Genet 2006; 70: 261–274.
Jacob KN, Garg A : Laminopathies: multisystem dystrophy syndromes. Mol Genet Metab 2006; 87: 289–302.
Youn G, Uzunyan M, Vachon L, Johnson J, Winder T, Yano S : Autosomal recessive LMNA mutation causing restrictive dermopathy. Clin Genet 2010; 78: 199–200.
Vytopil M, Benedetti S, Ricci E et al: Mutation analysis of the lamin A/C gene (LMNA) among patients with different cardiomuscular phenotypes. J Med Genet 2003; 40: e132–e132.
Morais P, Magina S, do Céu Ribeiro M et al: Restrictive dermopathy—a lethal congenital laminopathy. Case report and review of the literature. Eur J Pediatr 2009; 168: 1007–1012.
Simha V, Agarwal AK, Oral EA, Fryns J-P, Garg A : Genetic and phenotypic heterogeneity in patients with mandibuloacral dysplasia-associated lipodystrophy. J Clin Endocrinol Metab 2003; 88: 2821–2824.
Acknowledgements
We sincerely thank the patient and his family for their enthusiastic participation. We thank Drs Salma Wakil, Nada Altassan, Dorota Monies and Mohamed Aboelhouda from the genotyping, gene panel, sequencing and bioinformatics core facilities at KFSHRC for their technical help. This work was supported by the Saudi Human Genome Project and KACST grant 13-BIO1113-20 (FSA).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Sewairi, W., Assiri, A., Patel, N. et al. Distal acroosteolysis, poikiloderma and joint stiffness: a novel laminopathy?. Eur J Hum Genet 24, 1220–1222 (2016). https://doi.org/10.1038/ejhg.2015.265
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.265
This article is cited by
-
Is an association of acro-osteolysis, bone fragility, and enchondromatosis a newfound disease caused by an amplification of PTHLH? A case report
Pediatric Rheumatology (2022)
-
Lost bones: differential diagnosis of acro-osteolysis seen by the pediatric rheumatologist
Pediatric Rheumatology (2021)
-
Discovery of mutations for Mendelian disorders
Human Genetics (2016)
