
Abstract
The hereditary blistering disease junctional epidermolysis bullosa (JEB) is always accompanied by structural enamel abnormalities of primary and secondary dentition, characterized as amelogenesis imperfecta. Autosomal recessive mutations in LAMA3, LAMB3 and LAMC2 encoding the heterotrimer laminin 332 (LM-332) are among the genes causing JEB. While examining pedigrees of JEB patients with LAMA3 mutations, we observed that heterozygous carriers of functional null mutations displayed subtle enamel pitting in the absence of skin fragility or other JEB symptoms. Here, we report two new LAMA3 functional null mutations: nonsense c.2377C>T p.(Arg793Ter) and splice-site c.4684+1G>A mutation in heterozygous carriers exhibiting enamel pitting. Both parents had offspring affected with JEB and displayed subtle enamel pitting of secondary dentition without any sign of skin blistering. The reported enamel abnormality in LAMA3 mutation carriers could be attributed to a half dose effect of the laminin α3 chain (haploinsufficiency).
Similar content being viewed by others


Introduction
Epidermolysis bullosa (EB) is an inherited blistering disease affecting the adhesion of epithelial cells.1 Autosomal recessive (and very seldom dominant) mutations in the genes COL17A1, ITGA3, ITGA6, ITGB4, LAMA3, LAMB3 and LAMC2 cause the junctional subtype (JEB).1 The latter three genes encode the individual chains of the heterotrimer laminin 332 (LM-332).1 In our Dutch EB cohort, 60% of JEB cases are caused by mutations in the LM-332 genes, of which 28% are in LAMA3 (unpublished data). Clinically, patients with JEB exhibit skin and mucous membrane blistering, nail dystrophy, and structural enamel abnormalities termed amelogenesis imperfecta.2 Primary and secondary dentition are always affected in JEB.2 Enamel in JEB is thin, hypoplastic and contains a 20% lower mineral content than that of healthy individuals.3, 4 Recently, we reported that heterozygous carriers of loss of function (null) mutations in LAMA3 exhibit dental abnormalities without skin symptoms.5 Carriers exhibited localized enamel pitting and hypoplasia of secondary dentition. Here, we report two new heterozygous carriers affected with dental abnormalities from loss of function mutations in LAMA3.
Subjects and methods
We studied two kindreds of Dutch origin, of which the index patients were referred to our clinic with suspected EB. Assessment of skin fragility was based on review of patient history and specific anamnestic questions (ie, tolerating Band-Aids, wound healing) and dermatological physical examination. We harvested and processed skin specimens for immunofluorescence antigen mapping (IF) and transmission electron microscopy (TEM) of the JEB index patients as described before.6, 7 IF and TEM were not performed in carriers, as no biopsies were available. Genomic DNA was isolated from peripheral blood from the index patients and parents, and sequenced using Sanger’s sequencing by a candidate gene approach. Phenotype and variant information were submitted to Leiden Open Variant database http://dna2.leeds.ac.uk/LOVD/genes/LAMA3 (patient IDs 00000159 and 00000160). Immunoblotting of cell lysates from cultured patient keratinocytes (not carriers) was performed as described before.8 Dental examination of the index patients and parents was performed by an experienced maxillofacial dentist (CS), with more than 15 years of experience treating EB patients. Appropriate informed consent was obtained and patients were treated in accordance with the principles of the Declaration of Helsinki.
Results
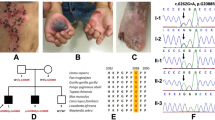
In Family 289, the index patient (EB-289) was a 2-year old boy and the third child of healthy, unrelated parents (Figure 1a). Beginning at 6 months of age, he began to develop blistering of hands, legs and ears together with nail dystrophy. He had generalized enamel pitting throughout his entire primary dentition, termed hypoplastic amelogenesis imperfecta (AI; Figure 1b). IF of intact skin showed normal expression of LM-332 (3+) when stained with the monoclonal antibody GB3 directed against the conformation epitope in the γ2 chain. Staining of the individual laminin α3 subunit with monoclonal antibody BM165 (gift from Dr Marinkovich, Stanford University) was decreased (2+) when compared with control (3+, Figure 1c). TEM of lesional skin on the left elbow confirmed cleavage through the lamina lucida fitting with JEB. Sequencing of DNA isolated from peripheral blood revealed that the patient was compound heterozygous for mutations in LAMA3 (NM_000227.4) c.(2377C>T);(4484C>T), concluding the diagnosis of JEB-generalized intermediate (previously JEB non-Herlitz).1 Additional screening of LAMB3 and COL17A1 revealed no mutations in the proband. We performed LAMA3 allele carrier analysis, and skin and dental examination in both parents. Exons are numbered like in NG_007853.2. The father (II-5) carried the novel c.(2377C>T); p.Arg793Ter mutation located in exon 19. In silico analysis of the mutation using AlamutVisual software (version 2.6.1, alamut.interactive-biosoftware.com) with protein and splice-site prediction programs for the paternal c.(2377C>T) mutation showed no alternate splicing, thus predicting a premature termination codon (PTC) p.(Arg793Ter). The father displayed subtle enamel pitting of secondary dentition (Figure 1b) and had no history of skin blistering. The mother (II-6) carried the missense mutation c.(4484C>T); p.(Ala1495Val) in exon 33, a mutation earlier reported in Yuen et al.5 Dental screening for pathology was negative in the mother, and dermatological examination was unremarkable. The parents reported that the index patient’s two older brothers (III-1 and III-2) had no skin disease or dental problems, but were unavailable for objective examination and mutation analysis.

Family 289 (a) Family pedigree with indicated genotypes of studied individuals (wt: wild type). (b) Generalized hypoplastic amelogenesis imperfecta in primary teeth of the index patient (III-3) His father (II-5) showed clinically silent localized enamel pitting on the cervical area of the lower canine as indicated by arrows. (c) IF antigen mapping of the α3 chain with BM165 showed 2+ expression when compared with 3+ in normal human skin (NHS) in the index patient.
The female child (EB-351, III-1) of Family 351 was seen in our clinic shortly after birth. The age of gestation was 35 weeks, and the parents were unrelated (Figure 2a). The child presented with skin fragility and absent nails of digits 1-4 of both hands. IF of lesional skin indicated strongly reduced staining of GB3 (1+) compared with control (3+). Antigen mapping showed GB3 exclusively in the blister roof and type VII collagen exclusively in the blister floor fitting a diagnosis of JEB. Additional staining with BM165 (Figure 2c) for the laminin α3 chain was strongly reduced (1+) when compared with healthy controls (3+). Genetic analysis revealed compound heterozygous mutations c.2910_2911delAA, in exon 22 and c.4684+1G>A in intron 34 of the LAMA3 gene. She was diagnosed with JEB-generalized severe (former JEB-Herlitz).1 DNA analysis of the father (II-1) revealed a heterozygous mutation in LAMA3: c.2910_2911delAA. The c.2910_2911delAA mutation caused a frameshift leading to a PTC, p.(Lys970AsnfsTer8). He was not available for clinical assessment. DNA analysis of the mother (II-2) revealed a heterozygous c.4684+1G>A mutation affecting the consensus donor splice site sequence of intron 34. Dental examination showed focal enamel pitting and grooves in some of her secondary dentition that had gone earlier unnoticed (Figure 2b). Skin fragility was absent. To further investigate the consequence of the maternal splice-site mutation on mRNA, we performed RT-PCR and subsequent sequencing of RNA isolated from the index’s keratinocytes. Three cDNA products of different length were identified. The shortest product resulted from skipping of exon 34 and 35 sequences (291 base pairs length) encoding an in-frame transcript. The second product showed skipping of exon 34 (160 base pairs) resulting in an out-of-frame transcript with introduction of a PTC. The third was a full-length wild-type transcript. Subsequently, we performed immunoblot in order to assess LM-332 protein expression using cell lysates of skin keratinocytes as described earlier.9 Staining with BM165 indicated a severely reduced amount of polypeptide of less than 2%, when compared with normal human control keratinocytes (Figure 2d). No smaller protein products were observed, suggesting that only the wild-type transcript from the maternal allele attributed to laminin-α3 expression.

Family 351 (a) Family pedigree. (b) Dental examination showing with arrows subtle enamel pitting on the labial surface of the lower canine by the mother (II-2) who carried the c.4684+1G>A mutation. Her upper and lower central and lateral incisors displayed the same focal pitting and grooves. (c) IF antigen mapping with BM165 showing severely diminished expression as compared with normal human skin (NHS). (d) Immunoblotting of cell lysates from cultured keratinocytes derived from healthy human skin (control) and from the index patient III-1. The α chain of LM-332 stained with monoclonal antibody BM165 exists in a processed (190 kDa) and non–processed (170 kDa) form. Patient extract (middle lane) compared with 1/4 diluted (left) and 1/64 diluted (right) extracts of control healthy keratinocytes, indicated that the amount of α chain expressed by the patient was <2% of normal. The blots were cropped for presentation clarity and alignment.
Discussion
Various degrees of defects of amelogenesis have been observed in mutation carriers of several JEB genes (COL17A1, LAMA3 and LAMB3, Table 1).2, 5, 10, 11, 12, 13, 14 In the examined heterozygous null carriers in both our families, dental pathology was localized focally, clinically asymptomatic and therefore not brought to attention until we examined JEB in their offspring. Since the first report of enamel abnormalities in LAMA3 null mutation carriers by Yuen et al.,5 we began to screen carriers of LAMA3 mutations for dental pitting. The low incidence of LAMA3 mutations, subtle nature of changes and lack of direct screening may explain the scarcity of reports of enamel defects in this population. In teeth, LM-332 is known to be a principal anchoring protein actively involved in the secretory stage of ameloblasts, an early stage of differentiation.10 Amelogenesis is a tightly regulated process in which ameloblasts express LM-332.3 Replacement of the lamina basale during amelogenesis is impaired if LM-332 is altered.3 Enamel and skin apparently respond differently to heterozygous mutations affecting the α chain, because skin fragility was absent in carriers. In teeth, a single allele defect in the α chain in null mutation carriers is enough to disrupt ameloblast-coordinated replacement of basement membrane macromolecules causing enamel pitting.4 The mechanism by which the heterozygous functional null mutations in LAMA3 result in disease appears to be haploinsufficiency, as we suggested before.5 In skin, keratinocyte adhesion is redundantly regulated, and if LM-332 is half dose, it is most likely sufficiently compensated for by other epidermal proteins, such as integrins.5, 15 The splice-site mutation c.4684+1G>A behaves like a LAMA3 null mutation, as <2% expression of intact laminin α3 protein was detected in the immunoblot (Figure 2d). The low amount of polypeptide can be explained by the detrimental effect on protein stability of the deleted amino acids in the G5 subdomain (codons 1509–1606) encoded by exons 34 and 35. Another possibility could be that the BM165 antibody recognizes an epitope for binding, which is missing from the internally truncated protein that is formed by this mRNA transcript. Although this cannot be completely excluded, it is highly unlikely, since the affected child in Family 351 had the most severe form of JEB due to almost complete loss of LM-332 and deceased at 6 months of age.
LAMA3 null mutation carriers show minor enamel changes, whereas carriers with LAMB3 mutations have deeper pits and visible grooves requiring medical attention.10, 11, 12 Extensive restorative dental care and multiple extractions such as described by Poulter et al. for LAMB3 (NM_000228.2) carriers were not present in our LAMA3 carriers. A difference in the reported cases is that in LAMB3 carriers, dental abnormalities are associated with mutations that are predicted to escape nonsense mediated RNA decay (NMD, Table 1). All mutations, except for one, were located in the last exon which predicted formation of a shortened laminin-β3 peptide truncated at the carboxy-terminus.12 The formation of abnormal truncated laminin β3 is more detrimental on enamel development than haploinsufficiency of laminin α3. Of note is that in a very recent publication two LAMB3 null mutations were described resulting in hypoplastic enamel.16 However, in this publication there was only a minor description of the phenotype, for the c.1903C>T (p.R635X) mutation there was no clear segregation with the dental phenotype, and for the c.124C>T mutation only one individual was affected with dental abnormalities. It yet remains to be determined whether the dental abnormalities were indeed due to the LAMB3 null mutation, or whether other mutations or factors were the underlying cause. For the COL17A1 gene both null mutations as well as one missense mutation have been described.
In conclusion, our observations confirm the hypothesis that carriers of functional null mutations in LAMA3 can exhibit subtle, localized enamel pitting due to haploinsufficiency. We advise dental screening of parents of JEB patients and siblings.
References
Fine JD, Bruckner-Tuderman L, Eady RA et al: Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol 2014; 70: 1103–1126.
McGrath JA, Gatalica B, Li K et al: Compound heterozygosity for a dominant glycine substitution and a recessive internal duplication mutation in the type XVII collagen gene results in junctional epidermolysis bullosa and abnormal dentition. Am J Pathol 1996; 148: 1787–1796.
Hu JC, Chun YH, Al Hazzazzi T, Simmer JP : Enamel formation and amelogenesis imperfecta. Cells Tissues Organs 2007; 186: 78–85.
Kirkham J, Robinson C, Strafford SM et al: The chemical composition of tooth enamel in junctional epidermolysis bullosa. Arch Oral Biol 2000; 45: 377–386.
Yuen WY, Pasmooij AM, Stellingsma C, Jonkman MF : Enamel defects in carriers of a novel LAMA3 mutation underlying epidermolysis bullosa. Acta Derm Venereol 2012; 92: 695–696.
Jonkman MF, de Jong MC, Heeres K et al: 180-kD bullous pemphigoid antigen (BP180) is deficient in generalized atrophic benign epidermolysis bullosa. J Clin Invest 1995; 95: 1345–1352.
Jonkman MF, de Jong MC, Heeres K, Sonnenberg A : Expression of integrin alpha 6 beta 4 in junctional epidermolysis bullosa. J Invest Dermatol 1992; 99: 489–496.
Pas HH, Kloosterhuis GJ, Heeres K et al: Bullous pemphigoid and linear IgA dermatosis sera recognize a similar 120-kDa keratinocyte collagenous glycoprotein with antigenic cross-reactivity to BP180. J Invest Dermatol 1997; 108: 423–429.
Gostynska KB, Nijenhuis M, Lemmink H et al: Mutation in exon 1a of PLEC, leading to disruption of plectin isoform 1a, causes autosomal-recessive skin-only epidermolysis bullosa simplex. Hum Mol Genet 2015; 24: 3155–3162.
Kim JW, Seymen F, Lee KE et al: LAMB3 mutations causing autosomal-dominant amelogenesis imperfecta. J Dent Res 2013; 92: 899–904.
Poulter JA, El-Sayed W, Shore RC et al: Whole-exome sequencing, without prior linkage, identifies a mutation in LAMB3 as a cause of dominant hypoplastic amelogenesis imperfecta. Eur J Hum Genet 2014; 22: 132–135.
Lee KE, Ko J, Le CG et al: Novel LAMB3 mutations cause non-syndromic amelogenesis imperfecta with variable expressivity. Clin Genet 2015; 87: 90–92.
Wang X, Zhao Y, Yang Y, Qin M : Novel ENAM and LAMB3 mutations in Chinese families with hypoplastic amelogenesis imperfecta. PLOS One 2015; 10: e0116514.
Murrell DF, Pasmooij AM, Pas HH et al: Retrospective diagnosis of fatal BP180-deficient non-Herlitz junctional epidermolysis bullosa suggested by immunofluorescence (IF) antigen-mapping of parental carriers bearing enamel defects. J Invest Dermatol 2007; 127: 1772–1775.
Poulter JA, Brookes SJ, Shore RC et al: A missense mutation in ITGB6 causes pitted hypomineralized amelogenesis imperfecta. Hum Mol Genet 2014; 23: 2189–2197.
Prasad MK, Geoffroy V, Vicaire S et al: A targeted next-generation sequencing assay for the molecular diagnosis of genetic disorders with orodental involvment. J Med Genet 2016; 53: 98–110.
Almaani N, Liu L, Dopping-Hepenstal PJ et al: Autosomal dominant junctional epidermolysis bullosa. Br J Dermatol 2009; 160: 1094–1097.
Yoshiba K, Yoshiba N, Aberdam D et al: Expression and localization of laminin-5 subunits during mouse tooth development. Dev Dyn 1998; 211: 164–176.
Bailleul-Forestier I, Molla M, Verloes A, Berdal A : The genetic basis of inherited anomalies of the teeth. Part 1: clinical and molecular aspects of non-syndromic dental disorders. Eur J Med Genet 2008; 51: 273–291.
Acknowledgements
The authors would like to thank the patients for their participation in this study. We wish to acknowledge the support of Stichting Vlinderkind (Dutch Butterfly Foundation).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Gostyńska, K., Yan Yuen, W., Pasmooij, A. et al. Carriers with functional null mutations in LAMA3 have localized enamel abnormalities due to haploinsufficiency. Eur J Hum Genet 25, 94–99 (2017). https://doi.org/10.1038/ejhg.2016.136
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2016.136
This article is cited by
-
Organoids from human tooth showing epithelial stemness phenotype and differentiation potential
Cellular and Molecular Life Sciences (2022)
-
LAMB3 mediates apoptotic, proliferative, invasive, and metastatic behaviors in pancreatic cancer by regulating the PI3K/Akt signaling pathway
Cell Death & Disease (2019)
