
Abstract
Post-transcriptional tRNA modifications are numerous and require a large set of highly conserved enzymes in humans and other organisms. In yeast, the loss of many modifications is tolerated under unstressed conditions; one exception is the N6-threonyl-carbamoyl-adenosine (t6A) modification, loss of which causes a severe growth phenotype. Here we aimed at a molecular diagnosis in a brother and sister from a consanguineous family who presented with global developmental delay, failure to thrive and a renal defect manifesting in proteinuria and hypomagnesemia. Using exome sequencing, the patients were found to be homozygous for the c.974G>A (p.(Arg325Gln)) variant of the KAE1 gene. KAE1 is a constituent of the KEOPS complex, a five-subunit complex that catalyzes the second biosynthetic step of t6A in the cytosol. The yeast KAE1 allele carrying the equivalent mutation did not rescue the t6A deficiency of the kae1Δ yeast strain as efficiently as the WT allele; furthermore, t6A levels quantified by LC-MS/MS were lower in the kae1Δ strain which was complemented by the mutation than in the kae1Δ strain, which was complemented by the WT allele. We conclude that homozygosity for c.974G>A (p.(Arg325Gln)) in KAE1 likely exerts its pathogenic effect by perturbing t6A synthesis, thereby interfering with global protein production. This is the first report of t6A biosynthesis defect in human. KAE1 joins the growing list of cytoplasmic tRNA modification enzymes, all associated with severe neurological disorders.
Similar content being viewed by others

Introduction
Post-transcriptional modifications of RNA are ubiquitous, and among cellular RNAs, tRNA are by far the most extensively modified. A typical eukaryotic tRNA contains more than 10 modifications of different types and more than 120 distinct chemical modifications of tRNA nucleotide bases and sugars are known to date.1, 2 A correspondingly large number of enzymes have been identified that catalyze the numerous post-transcriptional tRNA modification reactions and these enzymes are often highly conserved, catalyzing similar modifications in multiple domains of life (reviewed in El Yacoubi et al.3). In yeast, the absence of many tRNA modifications is tolerated under standard, unstressed growth conditions, though often resulting in error-prone translation. In only a few cases does the absence of tRNA modifications lead to severe growth phenotypes. One such example is the N6-threonyl-carbamoyl-adenosine (t6A) modification, a complex modification of adenosine located at position 37 (t6A37) next to the anticodon stem loop of many tRNAs that decode ANN codons. The absence of this modification is associated with a severe growth phenotype in yeast,4, 5 possibly due to the fact that t6A is a precursor in the synthesis of other hypermodified derivatives such as cyclic-t6A (ct6A), N6-methyl-t6A (m6t6A)6, 7 and 2-methylthio-t6A (ms2t6A) (reviewed in ref. 8). In mammals, the absence of ms2t6A37 in tRNA-Lys in pancreatic beta cells results in the reduction of glucose-stimulated proinsulin synthesis, linking t6A derivatives to diabetes.9, 10
We now report a biosynthetic defect of the t6A molecule in two patients suffering from global developmental delay, failure to thrive and a renal defect manifesting by proteinuria and hypomagnesemia.
Subjects and methods
Patients
The subjects of this study were two siblings, a female, patient II-1, and a male, II-2, the children of an Arab Muslim first cousin parents (Figure 1a). Their clinical data are presented below as well as in Table 1. Patient II-1 was born at term with a birth weight of 2600 g. Dysmorphic features were noted at birth, including upturned nose, antimongoloid slant, high-arched palate and thoracolumbar kyphosis. Her clinical course was characterized by failure to thrive due to impaired swallowing (at 4 years, her weight was 11 kg), acquired microcephaly, hypotonia and gross motor delay, with both comprehensive and expressive language skills being severely impaired. At 6 years, she was unable to sit unsupported and her vocabulary was confined to 5–10 words.

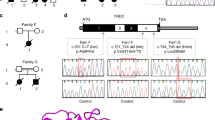
(a) Family pedigree and KAE1 genotype. The genotype of the c.974G>A, p.(Arg325Gln) mutation in the KAE1 gene is shown. Filled symbols denote patients. (b) The H. sapiens KAE1 Arg325 residue is conserved. Multiple alignments comparing Tcs3/Kae1 proteins from a diverse set of Eukarya and from Methanocaldoccus jannaschii with the mitochondrial homolog Tcs4/Qri7 that is closely related to the bacterial TsaD. The R325 residue (H. sapiens numbering) is conserved in all the KAE1 proteins but not in the QRI7 protein. (c) Chromatogram of part of exon 11 of KAE1 gene around the c.974 g>A mutation site (arrow) in the patient (upper panel), a carrier (middle panel) and a healthy control (lower panel). (d) Brain MRI of patient II-1 at 18 months of age. Left and center T2-weighted images showing periventricular high signal, right image showing isointense white and gray matter consistent with hypomyelination.
Brain MRI performed at the age of 4 years was reportedly normal except for cerebellar atrophy though the images were not available for review. The EEG was normal. Muscle biopsy disclosed type II atrophy.
Laboratory investigation revealed severe hypomagnesemia with normal calcium and albumin levels. A 24 h urine collection disclosed renal magnesium wasting combined with hypercalciuria and proteinuria. Normal serum magnesium levels were achieved with oral magnesium supplementation. The karyotype was 46XX and the metabolic screen was normal.
At 4 years, she was admitted with hypertensive crisis (blood pressure 120/180) that did not respond to intravenous hydralazine but normalized following intravenous β1-blocker (Esmolol) that was later replaced by oral propranolol and hydralazine. The cause of this event remained obscure as creatinine clearance, Doppler of renal arteries, blood levels of cortisol, ACTH, renin, aldosterone, PTH and thyroid parameters were all normal as was echocardiography. At the age of 6 years and 6 months, she was admitted with severe hypomagnesemia (0.1 mm, N=0.7–1.1), tachycardia and tachypnea. She improved on intravenous magnesium, but on the third day developed hypertensive crisis, which was refractory to treatment and led to her death.
Her brother, patient II-2 was born at term, with birth weight of 3800 g. Routine ultrasound examination during the pregnancy disclosed a hypoplastic left heart and aortic coarctation. At birth, physical examination revealed frontal bossing, bilateral microphthalmia, bilateral coloboma, bilateral ptosis, antimongoloid slant, strabismus, nystagmus, pectus excavatum and chest wall asymmetry, ichthyosis, arachnodactyly and cryptorchidism.
The patient underwent a cardiac surgical repair at 1 week of age and repair of pyloric stenosis at 4 months of age. Thereafter, the clinical course was characterized by failure to thrive, with microcephaly, nystagmus, hypotonia and gross motor delay, with both comprehensive and expressive language skills being severely impaired.
Brain MRI findings were consistent with leukodystrophy, showing increased T2 signal of white matter in a diffuse pattern (Figure 1d). EEG was normal. A normal 46XY karyotype was also obtained. A metabolic screening panel was normal.
Blood tests revealed severe hypomagnesemia with hypocalcemia. A 24 h urine collection disclosed renal magnesium wasting combined with hypercalciuria, which were successfully treated with oral magnesium and calcium supplements. The patient died of respiratory insufficiency in association with pneumonia at 8 years of age.
Methods
Chemicals and reagents
Benzonase, deferoxamine, butylated hydroxytoluene, acetonitrile and buffer salts were purchased from Sigma-Aldrich (St Louis, MO, USA). Calf intestine alkaline phosphatase was obtained from Thermo Fisher (Waltham, MA, USA). Coformycin was obtained from the US National Cancer Institute. Phosphodiesterase I was purchased from US Biological (Salem, MA, USA). Tetrahydrouridine was purchased from Calbiochem (San Diego, CA, USA). Sartorius Vivaspin 500-brand centrifugal filter units were used for dialysis and sample concentration.
Whole exome sequencing and bioinformatic analysis
Exonic sequences were enriched in the DNA samples of patients II-1 and II-2 using SureSelect Human All Exon 50 Mb Kit V4 (Agilent Technologies, Santa Clara, CA, USA). Sequences were determined by HiSeq2000 (Illumina, San Diego, CA, USA) as 100-bp paired-end runs. Data analysis including read alignment and variant calling was performed by DNAnexus software (Palo Alto, CA, USA) using the default parameters with the human genome assembly hg19 (GRCh37) as reference. Parental consent was given for DNA studies. The study was performed with the approval of the ethical committees of Hadassah Medical Center and the Ministry of Health.
The gene variant database is available at www.LOVD.nl/OSGEP (patient ID #00095419).
For homology comparison: individual sequences were obtained from the National Center for Biotechnology Information (NCBI) database (http://www.ncbi.nlm.nih.gov/books/NBK44864/). Multiple alignments were generated using MultiAlin,11 and the structures were visualized with the UCSF Chimera platform.12
Growth media
Escherichia coli was grown in Luria Bertani (LB) medium (Fisher Scientific) at 37 °C or on LB agar supplemented with ampicillin (Amp; 100 μg/ml); arabinose (Ara; 0.2%) or anhydrotetracycline (aTet; 250 ng/ml) were added as needed. The Escherichia coli Top10 strain (Invitrogen, Carlsbad, CA, USA) was routinely used for cloning.
Saccharomyces cerevisiae strains were routinely grown on YPD (DIFCO Laboratories) supplemented or not with G148 (0.2 mg/ml) at 30 °C unless otherwise stated. Synthetic minimal, with or without Agar, SD base or SD base Gal/Raf without dropout (−uracil) were purchased from Clontech (Palo Alto, CA, USA) and prepared as recommended by the manufacturer.
Complementing the E. coli tsaD and tsaC essentiality phenotypes by co-expressing the human Tcs1/Tcs4 genes
Homo sapiens Tcs1 and Tcs4 cDNA sequences were obtained from the NCBI database. An E. coli codon-optimized synthetic gene operon of Tcs1–Tcs4 (sequence given in Supplementary Data) was purchased from Genscript and cloned in a pBAD24 plasmid13 cut with NcoI/SphI d. The resulting plasmid (pBAD24-Tcs1/4Hs) or the empty pBAD24 vector were used to transform E. coli strains VDC5684 (BW25113 PTET:tsaC) and VDC5801 (BW25113 PTET:tsaD).14 Transformants were selected on LB agar supplemented with Amp and aTet.
Construction of pKae1 R376Q and complementation assays
Base changes leading to the R376Q mutation were introduced in the yeast KAE1/TCS2 gene cloned in the pYES derivative pKae14 by site-directed mutagenesis using the Q5 site-directed mutagenesis kit (New England Biolabs, NJ, USA) with the primers Fwd kae1 R376Q 5′-gcagaaattccaaaccgatgaag-3′ and RV kae1 5′-gtaacaacagtttcagaaaagtcct-3′ to give plasmid pKAE1_R376Q. The sequence of the insert was verified by Sanger DNA Sequencing (Eton Bioscience, San Diego, CA, USA).
For KAE1 complementation assays, the S. cerevisiae BY4741 kae1Δ strain (VDC5653)4 was transformed with 1 μg of plasmid pKAE1 or pKAE1_R376Q as described in the Zymo Research handbook (Zymo Research, Irvine, CA, USA), then recovered with 2 ml of YPD and placed at 30 °C for 2 h with shaking. Then the cells were pelleted by centrifugation, washed with 2 ml of water and resuspended in 100 μl of water before plating onto SD−uracil supplemented with G148 (0.2 mg/ml).
Isolated colonies of transformed cells were inoculated in 15 ml of SD−uracil supplemented with G148 (0.2 mg/ml) then diluted to an OD600=0.009 in 1 l of SD base Gal/Raf without uracil, G148 (0.2 mg/ml) and grown at 30 °C with shaking until they reach OD600=0.5. The experiment was performed with two biological replicates.
Extraction of bulk tRNA
Bulk tRNA were prepared as previously described.5 Briefly, tRNAs were extracted using acid-buffered phenol (saturated with 50 mm sodium acetate, pH 5.5) followed by ethanol precipitation. tRNA quality was evaluated by both 12% acrylamide, 8.0 m urea denaturing gel electrophoresis with ethidium bromide visualization and Bioanalyzer (Agilent Technologies) using a small RNA chip.
Positive hybridization in absence of t6A (PHAt6A) assay
Three volumes of denaturing solution (500 μl formamide, 162 μl 37% formaldehyde, 100 μl RNase-free 10X MOPS solution) was added to 1 μl of 1 ng/μl tRNA followed by tRNA denaturation at 85 °C for 15 min. The mixture was cooled on ice and the final volume adjusted to 30 μl with 20X SSC (1.5 m sodium chloride, 0.15 m sodium citrate). The mixture was spotted on Biodyne A membrane (Thermo Scientific, Waltham, MA, USA) and crosslinked under UV at 120 mJ/cm2. Prehybridization was performed by incubation in DIG Easy Hyb (Roche, Basel, Switzerland) for 60 min at 42 °C with constant rocking. Biotinylated DNA probes were designed to complement the tRNAIleIAU anticodon stem loop (5′(biosg)-CCCCGCGTTATTAGCAC-3′) and TΨC loop (5′(biosg)-GCGGGATCGAACCGC-3′). DNA probes (1 μl per 5 cm2 of membrane) were denatured in DIG Easy Hyb at 95 °C for 10 min before incubation with the membrane overnight at 40 °C with constant rocking. Membranes were washed three times with 10X SSC for 10 min at ambient temperature. The membrane hybridized with the anticodon stem loop (ASL) probe was subjected to two extra washes, one at ambient temperature and another at 42 °C. Detection and visualization was performed with North2South Chemiluminescent Detection Kit, as described by the manufacturer (Thermo Scientific).
Mass spectrometric analysis of t6A level
Two micrograms of bulk tRNA from each sample were hydrolyzed to ribonucleosides in a reaction containing Benzonase (0.375 U), calf intestine alkaline phosphatase (8.5 U), phosphodiesterase I (0.05 U), coformycin (3.5 μm; nucleobase deaminase inhibitor), deferoxamine (3 mm; antioxidant), butylated hydroxytoluene (0.3 mm; antioxidant) and MgCl2 (5 mm) in a final reaction volume of 50 μl. The reaction was allowed to proceed for 2 h at 37 °C and was stopped by removal of the enzymes by microfiltration with 10 000 Da spin filters. Following the addition of [15N5]-2’-deoxyadenosine as an internal standard for data normalization, ribonucleosides were resolved on a Synergy Fusion RP HPLC column (2.5 μm particle size, 100 Å pore size, 100 mm length, 2 mm inner diameter; Phenomenex, Torrance, CA, USA) mounted on an Agilent 1290 series HPLC system equipped with a diode array detector (DAD). The ribonucleosides were eluted at a flow rate of 0.35 ml/min and a column temperature of 35 °C with a gradient consisting of 5 mm ammonium acetate (A) and acetonitrile (B) as follows: 0–1 min 100% A, 1–10 min 0–10% B, 10–24 min 10–40% B, 24–44 min 40–80% B, and 44–49 min 100% A to regenerate the column. The column, with its eluent directed through the DAD to record the 260 nm absorbance of canonical ribonucleosides, was coupled to an Agilent 6430 triple quadrupole mass spectrometer operated in positive ion mode with the following parameters: electro-spray ionization (ESI-MS), fragmentor voltage (average) 80 V, cell accelerator voltage 2 V, N2-gas temperature 350 °C, N2-gas flow 10 l/min, nebulizer 40 p.s.i., capillary 3500 V. Using dynamic multiple reaction monitoring (MRM), modified ribonucleosides were identified based on retention time (t6A at 7.9–8.3 min) and mass transition (m/z 413→281 for loss of ribose from t6A). The signal for t6A was normalized by dividing by the peak area of the [15N5]-dA standard (inter-sample variation) and by the summed MRM peak areas of the canonical ribonucleosides (input RNA variation). The normalized peak areas of two biological replicates were then averaged.
Results
Exome analysis of patients II-1 and II-2 yielded 57.0 and 71.3 million mapped reads with a mean coverage of X83 and X111, respectively. Following alignment to the reference genome (Hg19) and variant calling, we performed coverage analysis, which was normal in patient II-1 but disclosed a duplication of hg19 Chr1: g.146638410_ 147380068dup in patient II-2 sample. The duplication included the genes PRKAB2, FMO5, CHD1L, BCL9, ACP6 and GJA5. Gains at 1q21.1, which span these genes, were previously reported to be significantly enriched among patients with congenital heart defects including tetralogy of Fallot, coarctation of the aorta and Shone's syndrome. These malformations were present even when the duplicated region was only 0.4 Mb long and included only PRKAB2, FMO5 and CHD1L.15, 16 We propose that the hypoplastic left heart and aortic coarctation, microphthalmia, bilateral coloboma, ichthyosis and arachnodactyly are all resulting from the Chr1q21.1 duplication.
Using the patients' variant lists, we performed a series of filtering steps under the hypothesis of a recessively inherited, homozygous, rare, causal allele. These included removing variants that were not shared by both patients, called less than X8, were off-target, heterozygous, synonymous, on the X chromosome, had MAF >1% at ExAC (Exome Aggregation Consortium, Cambridge, MA; URL: http://exac.broadinstitute.org) or MAF >4% at the Hadassah in-house database. Eleven homozygous variants remained (Supplementary Table S1), but, after genotyping the healthy sister and removing rare variants that were present in ExAC in homozygosity, we focused on hg19 chr14:g.20915433C>T, NM_017807.3:c.974G>A, p.(Arg325Gln; R325Q) in KAE1 (OSGEP). This variant affected an evolutionary conserved residue (Figure 1b), segregated in the family (Figure 1c), was carried by only 2 of the 60 700 individuals whose exome analyses were deposited at ExAC (accessed July 2016), and was not present in our in-house database (~900 Moslem-Arab exome analyses).
KAE1 encodes the TCS3/OSGEP/Kae1 protein that is a constituent of the KEOPS (Kinase, Endopeptidase and Other Proteins of small Size) complex. KEOPS complex participates in the synthesis of t6A, a modification commonly occurring in many tRNAs at position A37 (see Thiaville et al.,17 for review). A reconstruction of t6A synthesis in H. sapiens based on similarity with experimentally validated enzymes is shown Figure 2. t6A biosynthesis starts with the generation of threonylcarbamoyladenylate (TC-AMP) from ATP, threonine and C02, which is catalyzed by Tcs1/YrdC or Tcs2/Sua5.18, 19, 20 The KEOPS complex catalyzes the second biosynthetic step in the cytosol, the transfer of the carboxythreonyl moiety from the TC-AMP intermediate to the adenosine on the target tRNA, with KAE1 catalyzing this activity in humans.

Predicted synthesis pathway for the t6A modification and its derivatives in H. sapiens. This pathway was predicted from sequence similarity and has been experimentally validated by E. coli complementation (red; see also Supplementary Figure S2), in vitro experiments (purple) or in vivo experiments in human cells (green). Ref_seq identifiers and synonyms are listed here: Tcs1/YrdC=NP_078916.3; Tcs3/Kae1/OSGEP=NP_060277.1; Tcs4/Qri7/OSGEPL1=NP_071748.2; Tcs5/Bud32/PRPK=NP_291028.3; Tcs6/Cgi121/TPRKB=NP_057142.1; Tcs7/Pcc1/LAGE3=NP_006005.2; Tcs8/Gon7/C14orf142=NP_115879.2; TrmO= NP_057565.3; CDKAL1/MtaB=NP_060244.2. The full colour version of this figure is available at European Journal of Human Genetics online.
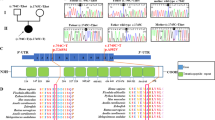
To examine the highly conserved Arg325 residue in the context of the entire KAE1 protein, we used the crystal structure of Methanocaldococcus jannaschii (Mj) Tcs3/Kae1 in complex with Tcs5/Bud32.21 In this structure (PDB: 3ENH), the conserved Arg (R315) is located at the periphery of the major binding interface between Tcs3/Kae1 and Tcs5/Bud32 (highlighted in red); the R315 side chain is displayed in the M. jannaschii protein structure in Supplementary Figure S1. The residue does not appear to participate in binding with Tcs5/Bud32, though mutations of residues in its proximity have been shown to present moderate growth defects.21 The Mj Kae1-Bud32 complex is exceptional because the two genes kae1 and bud32 are fused together and code for a single polypeptide. Consequently, the interaction surface (and the position of R315) between the two domains may be significantly different compared with human orthologs. Further, R315 is found in a signature motif that separates the Tcs3/Kae1 subfamily from the other members of the ASKHA ATPase family.21 Interestingly, this region shows some dynamics in folding depending on the context, as observed when comparing the structures of Kae1/Bud32 fusion alone (PDB: 3EN9) to those in complex with Bud32/Cgi121 (PDB: 3ENH). We observed that R315 is contained in a loop in PDB:3ENH while the residue is in a helical structure in PDB:3EN9. Further biochemical and structural studies are required to fully understand the functional importance of this strongly conserved residue.
To demonstrate that the KAE1 R325Q variant affects t6A synthesis, we took advantage of the strong homology between human and yeast KAE1 protein (Figure 1b). We and others have previously shown that tRNAs extracted from kae1Δ yeast strains are devoid of t6A and that this phenotype is partially complemented by expressing the WT KAE1 gene in trans on a plasmid (pKAE1). The position equivalent to R325 in the S. cerevisiae enzyme is R376 (Figure 1b). A pKae1 derivative harboring the R376Q mutation (pKAE1_R376Q) was constructed as described in the ‘Methods’ section and tested for complementation of the t6A deficiency phenotype of the S. cerevisiae kae1Δ strain. A t6A hybridization assay that produces a signal only in the absence of t6A22 was first used. Under specific stringency conditions, an oligonucleotide targeted to the anticodon stem loop (ASL) region of tRNAIleIAU binds only when the t6A modification is absent, such as in the kae1Δ strain (Figure 3a, left panel). This binding was totally eliminated when the WT KAE1 gene was expressed in trans but only partially eliminated when the mutant allele (KAE1_R376Q) was expressed (Figure 3a, right panel). These findings suggested that pKAE1_R376Q is unable to fully rescue the t6A synthesis defect of kae1Δ yeast strain. To further validate this result, t6A levels were quantified by liquid chromatography-coupled tandem mass spectrometry (LC-MS/MS). As shown in Figure 3b, tRNA extracted from the kae1Δ mutant transformed with the pKAE1_R376Q plasmid contained approximately 25% less t6A than the one transformed with the WT control. As a control for the fold-change analysis, the abundance ratios of the canonical ribonucleosides remained close to 1 as expected. Normalized levels of t6A in both sets of complementation samples were similar (well within one standard deviation of the mean for the two biological replicates). Of note, the pKAE1_R376Q plasmid was found to regularly revert to the WT sequence in the course of the experiment, so the reduction in t6A synthesis reported here might be an underestimate.

Ribonucleoside quantification in tRNA extracted from S. cerevisiae Δkae1 transformed with plasmids expressing the wild-type (WT) yeast KAE1 gene (pKAE1) or the derivative carrying the R376Q mutation (pKAE1 R376Q). (a) Northern dot-blot assay for the detection of t6A in bulk tRNA preparations. Hybridization with a probe against the tRNAIleIAU ASL shows a signal only when t6A is absent. The probe specific to the TΨC loop is used to measure the quantity of tRNA. Lanes with sua5Δ (catalyzes threonylcarbamoyladenylate synthesis) and kae1Δ serve as controls to demonstrate the lack of t6A synthesis. (b) LC-MS/MS analysis of t6A in bulk tRNA preparations. The fold-change values for complementation of the mutant compared with the WT alleles were calculated and averaged. The data represent mean±error about the mean for two biological replicates.
Discussion
The identification of disease-associated genes in patients with primary neuronal degenerative disorders was recently accelerated, thanks to the introduction of exome analysis. The major challenge for this platform is rare variant filtering, which is markedly alleviated when the patient is the product of a consanguineous marriage, as only homozygous variants are retained. Consanguinity is practiced by approximately 20% of the human population and amounts to 31% among Israeli Arabs23 and 40% among Palestinian Arabs.24 Since 2011, we have performed 1800 exome analyses, of which ~1500 were singleton. Of the total, 67% of the patients originated from consanguineous marriages and ~30% suffered from a primary neuronal degenerative disorder. Here we present genetic and molecular evidence for a mechanistic link between a variant KAE1 gene, defects in t6A synthesis and a global developmental and metabolic disorder in a brother and sister from a consanguineous family. The combination of brain and renal disease is relatively rare and the list of differential diagnosis includes defects of mitochondrial coenzyme Q biosynthesis,25, 26 mitochondrial translation defect due to SARS2 mutations,27 Galloway–Mowat syndrome due to WDR37 mutations28 and EAST syndrome due to KCNJ10 mutations.29 The specific renal tubular reabsorption defect of the divalent ions magnesium and calcium observed in our patients is characteristic of hypercalciuric hypomagnesemic syndromes. The underlying molecular basis of this group of disorders are mutations in genes that are mainly expressed in the thick ascending loop of Henle in the kidney, CLDN16 and CLDN19, encoding tight junction proteins.30, 31 However, brain involvement was not reported in these patients, which suggests that our patients suffer from a novel syndrome. Using exome analysis, we identified a homozygous c.974G>A (p.(Arg325Gln)) mutation in the KAE1 gene. The mutation segregated with the disease in the family, was extremely rare, and affected a highly conserved residue in the KAE1 protein. As noted earlier, KAE1 is part of the KEOPS complex that catalyzes the second biosynthetic step of t6A in the cytosol, the transfer of the carboxythreonyl moiety from the TC-AMP intermediate to the adenosine on the target tRNA. The same reaction is catalyzed by a unique protein, Tcs4/Qri7/OSGEPL1, in the mitochondria.32, 33 The t6A biosynthetic enzymes are conserved throughout evolution and we were able to complement the essentiality phenotypes caused by depletion of tsaC (the E. coli Tcs1 ortholog) or tsaD (the E. coli Qri7 ortholog) in E. coli strains by co-expressing the human genes Tcs1 and Tcs4 in a pBAD24 derivative in trans (Supplementary Figure S2).
A recent study has shown that the human KEOPS complex is composed of the five subunits TCS3/OSGEP/Kae1, TCS5/PRPK/Bud32, TCS6/LAGE3/Pcc1, TCS7/TPRKB/Cgi121 and Tcs8/C14ORF142/Gon7, which catalyze the t6A formation from TC-AMP in vitro.34 Genes involved in several hypermodification derivatives of t6A are also conserved in H. sapiens (Figure 2). The function of the human TrmO protein in m6t6A synthesis and MtaB proteins in ms2t6A synthesis have been validated in vitro.7, 35 In addition, the role of MtaB/CDKAL1 has been confirmed in vivo in mouse and humans.9 ct6A has not been found in human cells6 and no homologs to the yeast or bacterial ct6A enzymes are found in the human genome (https://genome.ucsc.edu). Collectively these results strongly suggest that most human t6A synthesis genes have been identified, though final validation awaits gene depletion in human cells.
The presence of this set of enzymes in all species attests to the importance of t6A in cell physiology. Although S. cerevisiae kae1Δ strains are viable, they have severe growth defects and pleiotropic phenotypes4, 18 and this is also the case in another fungus, Aspergillus nidulans.36 In the human malaria parasite Plasmodium falciparum, the cytoplasmic KAE1 gene appears to be essential.37 Recent studies have shown that the absence of KAE1 also leads to strong phenotypes in multicellular organisms, such tissue-specific cell growth regulated by t6A in flies.38, 39 It is thus not surprising that a mutation that lowers t6A levels in humans even by 25% would lead to severe clinical phenotype, raising the possibility that Kae1 null could be embryonic lethal.
Human diseases associated with defects in tRNA modification are mainly affecting mitochondrial tRNAs (reviewed in Powell et al.39). However, defects in cytoplasmic tRNA modifications are increasingly recognized as a cause of human diseases, most commonly involving the brain, as in PUS3, ADAT3, WDR4, NSUN2 and FTSJ1 mutations.40, 41, 42, 43, 44 Additional organ systems are less frequently affected in cytosolic tRNA modification defects, such as pancreatic beta cells in TRMT10A mutations45 and, in the present report, a KAE1 mutation that affects renal tubuli.
In summary, homozygosity for R325Q in the KAE1 gene is associated with global developmental delay, failure to thrive, and a renal tubular defect. The mutation appears to exert its pathogenic effects by perturbing t6A synthesis, possibly interfering with global protein synthesis. KAE1 is the first reported t6A biosynthesis defect in humans and joins the growing list of cytoplasmic tRNA modification enzymes associated with severe neurological disorders.
References
Machnicka MA, Milanowska K, Osman Oglu O et al: MODOMICS: a database of RNA modification pathways: 2012 update. Nucleic Acids Res 2013; 41: D262–D267.
Jackman JE, Alfonzo JD : Transfer RNA Modifications: nature's combinatorial chemistry playground. Wiley Interdiscip Rev RNA 2013; 4: 35–48.
El Yacoubi B, Bailly M, de Crécy-Lagard V : Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu Rev Genet 2012; 46: 69–95.
El Yacoubi B, Hatin I, Deutsch C et al: A role for the universal Kae1/Qri7/YgjD (COG0533) family in tRNA modification. EMBO J 2011; 30: 882–893.
El Yacoubi B, Lyons B, Cruz Y et al: The universal YrdC/Sua5 family is required for the formation of threonylcarbamoyladenosine in tRNA. Nucleic Acids Res 2009; 37: 2894–2909.
Miyauchi K, Kimura S, Suzuki T : A cyclic form of N6-threonylcarbamoyladenosine as a widely distributed tRNA hypermodification. Nat Chem Biol 2013; 9: 105–111.
Kimura S, Miyauchi K, Ikeuchi Y, Thiaville PC, de Crécy-Lagard V, Suzuki T : Discovery of the β-barrel-type RNA methyltransferase responsible for N6-methylation of N6-threonylcarbamoyladenosine in tRNAs. Nucleic Acids Res 2014; 42: 9350–9365.
Sarin LP, Leidel SA : Modify or die?—RNA modification defects in metazoans. RNA Biol 2014; 11: 1555–1567.
Wei FY, Suzuki T, Watanabe S et al: Deficit of tRNA(Lys) modification by Cdkal1 causes the development of type 2 diabetes in mice. J Clin Invest 2011; 121: 3598–3608.
Wei FY, Tomizawa K : Functional loss of Cdkal1, a novel tRNA modification enzyme, causes the development of type 2 diabetes. Endocr J 2011; 58: 819–825.
Corpet F : Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res 1988; 16: 10881–10890.
Pettersen EF, Goddard TD, Huang CC et al: UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 2004; 25: 1605–1612.
Guzman LM, Belin D, Carson MJ, Beckwith J : Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 1995; 177: 4121–4130.
Thiaville PC, El Yacoubi B, Köhrer C et al: Essentiality of threonylcarbamoyladenosine (t(6)A), a universal tRNA modification, in bacteria. Mol Microbiol 2015; 98: 1199–1221.
Tomita-Mitchell A, Mahnke DK, Struble CA et al: Human gene copy number spectra analysis in congenital heart malformations. Physiol Genomics 2012; 44: 518–541.
Greenway SC, Pereira AC, Lin JC et al: De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat Genet 2009; 41: 931–935.
Thiaville PC, Iwata-Reuyl D, de Crécy-Lagard V : Diversity of the biosynthesis pathway for threonylcarbamoyladenosine (t(6)A), a universal modification of tRNA. RNA Biol 2014; 11: 1529–1539.
Thiaville PC, Legendre R, Rojas-Benítez D et al: Global translational impacts of the loss of the tRNA modification t6A in yeast. Microb Cell 2016; 3: 29–45.
Perrochia L, Crozat E, Hecker A et al: In vitro biosynthesis of a universal t6A tRNA modification in Archaea and Eukarya. Nucleic Acids Res 2013; 41: 1953–1964.
Perrochia L, Guetta D, Hecker A, Forterre P, Basta T : Functional assignment of KEOPS/EKC complex subunits in the biosynthesis of the universal t6A tRNA modification. Nucleic Acids Res 2013; 41: 9484–9499.
Mao DY, Neculai D, Downey M et al: Atomic structure of the KEOPS complex: an ancient protein kinase-containing molecular machine. Mol Cell 2008; 32: 259–275.
Rojas-Benitez D, Thiaville PC, de Crécy-Lagard V, Glavic A : The levels of a universally conserved tRNA modification regulate cell growth. J Biol Chem 2015; 290: 18699–18707.
Sharkia R, Mahajnah M, Athamny E, Khatib M, Sheikh-Muhammad A, Zalan A : Changes in marriage patterns among the Arab community in Israel over a 60-year period. J Biosoc Sci 2016; 48: 283–287.
Sirdah MM : Consanguinity profile in the Gaza Strip of Palestine: large-scale community-based study. Eur J Med Genet 2014; 57: 90–94.
Lopez LC, Schuelke M, Quinzii CM et al: Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet 2006; 79: 1125–1129.
Diomedi-Camassei F, Di Giandomenico S, Santorelli FM et al: COQ2 Nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol 2007; 18: 2773–2780.
Belostotsky R, Ben-Shalom E, Rinat C et al: Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am J Hum Genet 2011; 88: 193–200.
Colin E, Huynh Cong E, Mollet G et al: Loss-of-function mutations in WDR73 are responsible for microcephaly and steroid-resistant nephrotic syndrome: Galloway-Mowat syndrome. Am J Hum Genet 2014; 95: 637–648.
Bockenhauer D, Feather S, Stanescu HC et al: Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med 2009; 360: 1960–1970.
Müller D, Kausalya PJ, Claverie-Martin F et al: A novel claudin 16 mutation associated with childhood hypercalciuria abolishes binding to ZO-1 and results in lysosomal mistargeting. Am J Hum Genet 2003; 73: 1293–1301.
Konrad M, Schaller A, Seelow D et al: Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet 2006; 79: 949–957.
Thiaville PC, El Yacoubi B, Perrochia L et al: Cross kingdom functional conservation of the core universally conserved threonylcarbamoyladenosine tRNA synthesis enzymes. Eukaryot Cell 2014; 13: 1222–1231.
Wan LC, Mao DY, Neculai D et al: Reconstitution and characterization of eukaryotic N6-threonylcarbamoylation of tRNA using a minimal enzyme system. Nucleic Acids Res 2013; 41: 6332–6346.
Wan L, Maisonneuve P, Szilard R et al: Proteomic analysis of the human KEOPS complex identifies C14ORF142 as a core subunit homologous to yeast Gon7. Nucleic Acids Res 2017; 45: 805–817.
Arragain S, Handelman SK, Forouhar F et al: Identification of eukaryotic and prokaryotic methylthiotransferase for biosynthesis of 2-methylthio-N6-threonylcarbamoyladenosine in tRNA. J Biol Chem 2010; 285: 28425–28433.
Dzikowska A, Grzelak A, Gawlik J et al: KAEA (SUDPRO), a member of the ubiquitous KEOPS/EKC protein complex, regulates the arginine catabolic pathway and the expression of several other genes in Aspergillus nidulans. Gene 2015; 573: 310–320.
Mallari JP, Oksman A, Vaupel B, Goldberg DE : Kinase-associated endopeptidase 1 (Kae1) participates in an atypical ribosome-associated complex in the apicoplast of Plasmodium falciparum. J Biol Chem 2014; 289: 30025–30039.
Lin CJ, Smibert P, Zhao X et al: An extensive allelic series of Drosophila kae1 mutants reveals diverse and tissue-specific requirements for t6A biogenesis. RNA 2015; 21: 2103–2118.
Powell CA, Nicholls TJ, Minczuk M : Nuclear-encoded factors involved in post-transcriptional processing and modification of mitochondrial tRNAs in human disease. Front Genet 2015; 6: 79.
Shaheen R, Han L, Faqeih E et al: A homozygous truncating mutation in PUS3 expands the role of tRNA modification in normal cognition. Hum Genet 2016; 135: 707–713.
Alazami AM, Hijazi H, Al-Dosari MS et al: Mutation in ADAT3, encoding adenosine deaminase acting on transfer RNA, causes intellectual disability and strabismus. J Med Genet 2013; 50: 425–430.
Shaheen R, Abdel-Salam GM, Guy MP et al: Mutation in WDR4 impairs tRNA m7G46 methylation and causes a distinct form of microcephalic primordial dwarfism. Genome Biol 2015; 16: 1–11.
Abbasi-Moheb L, Mertel S, Gonsior M et al: Mutations in NSUN2 cause autosomal-recessive intellectual disability. Am J Hum Genet 2012; 90: 847–855.
Ramser J, Winnepenninckx B, Lenski C et al: A splice site mutation in the methyltransferase gene FTSJ1 in Xp11.23 is associated with non-syndromic mental retardation in a large Belgian family (MRX9). J Med Genet 2004; 41: 679–683.
Gillis D, Krishnamohan A, Yaacov B, Shaag A, Jackman JE, Elpeleg O : TRMT10A dysfunction is associated with abnormalities in glucose homeostasis, short stature and microcephaly. J Med Genet 2014; 51: 581–586.
Acknowledgements
We thank Sean Beckman for technical help. This work was supported by the National Institutes of Health (grant number R01 GM70641 to VdC-L), by the Singapore-MIT Alliance for Research and Technology under a grant from the National Research Foundation of Singapore (to PCD) and by the Trudy Mandel Louis Charitable Trust (to OE).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Edvardson, S., Prunetti, L., Arraf, A. et al. tRNA N6-adenosine threonylcarbamoyltransferase defect due to KAE1/TCS3 (OSGEP) mutation manifest by neurodegeneration and renal tubulopathy. Eur J Hum Genet 25, 545–551 (2017). https://doi.org/10.1038/ejhg.2017.30
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2017.30
This article is cited by
-
Whole-genome resequencing provides insights into the diversity and adaptation to desert environment in Xinjiang Mongolian cattle
BMC Genomics (2024)
-
Diagnosis delay a family of Galloway-Mowat Syndrome caused by a classical splicing mutation of Lage3
BMC Nephrology (2023)
-
Crystal structure of the human PRPK–TPRKB complex
Communications Biology (2021)
-
The expanding world of tRNA modifications and their disease relevance
Nature Reviews Molecular Cell Biology (2021)
-
Modifications of the human tRNA anticodon loop and their associations with genetic diseases
Cellular and Molecular Life Sciences (2021)

{kind=link}