
Abstract
Aim
To investigate and correlate the frequency and types of pupil abnormalities that are associated with hereditary peripheral neuropathy in a large cohort of patients prospectively examined.
Methods
A prospective study between 1998 and 2007. Patients were enrolled and examined after being seen in the neurology clinic. Data were collected on demographics, family and medical history. Patients had eye and pupillography testing carried out as well as being neurologically and genetically investigated.
Results
A consecutive series of 131 cases of inherited peripheral neuropathy were seen and categorized into five groups: familial amyloid polyneuropathy (FAP), Charcot Marie Tooth disease (CMT), hereditary neuropathywith liability to pressure palsies (HNPP), Refsum's disease, and hereditary sensory and autonomic neuropathy. A number of unreported mutations were identified in these patient groups. Pupil abnormalities were common in the Refsum's group, with frequent abnormally small pupils. The inherited neuropathies commonly associated with autonomic abnormalities were frequently found to have developed bilateral Horner's syndrome, which was particularly prevalent in our FAP series. Abnormalities were rare in HNPP and CMT type 1, but CMT type 2 showed frequent and varied pupil defects. The results describe the pupil abnormalities that were frequently associated with the particular group of inherited neuropathy patients, but we could not predict the genetic defect or the neuropathy severity.
Conclusions
This is the first study of the pupil abnormalities found in the inherited neuropathies and provides an overview of the frequency and type of defects seen in a large number of cases. This series along with the detailed tables will act as an important diagnostic aid in assessing these patients.
Similar content being viewed by others

Introduction
Inherited neuropathies are common disorders with a prevalence of at least 1 in 2500 individuals.1 The classification is based on clinical features and electrophysiology, but over the last few years, genetic testing has shown that even clinically identical inherited neuropathies can be caused by different genetic defects.2, 3, 4, 5, 6, 7, 8 Pupil abnormalities are an important aspect of the clinical characterization of patients with inherited neuropathy, but they have been investigated only in isolated cases or small groups.9 Please refer to the Supplementary text, which has an extensive review of the literature on pupil abnormalities, as only brief background information is given here.
Familial amyloid polyneuropathy (FAP) is an inherited neuropathy with autonomic and systemic manifestations.10 In FAP, there is deposition of amyloid material within the autonomic nerve supply of the ocular tissues, predominantly the vitreous body, conjunctivae, and ciliary ganglia,11 with the development of scalloped pupils.12, 13 The pupil abnormalities in FAP have also been identified in immunoglobulin light-chain-associated amyloidosis,14 but only a small number of the commonest Transthyretin FAP mutation types have been investigated.14, 15, 16 Abnormal ocular signs are also an important feature of Refsum's disease as in Refsum's original report17 where the pupils were miotic and responded poorly to light, better to near, and dilated poorly in response to homatropine eyedrops. Similar observations have been made by subsequent authors,18 but only in single cases or small series. The neurological phenotype of hereditary sensory and autonomic neuropathy (HSAN) is similar to FAP, but there have been no cases or families reported examined in detail.19
In Charcot Marie Tooth disease (CMT), pupil abnormalities have been reported over the years but mainly in single families,9 and most cases were diagnosed with the clinically severe form of CMT called Déjérine–Sottas syndrome. Recently, pupil abnormalities have been shown to be particularly prevalent in certain myelin protein zero (MPZ) gene mutations,20, 21 and sluggish pupillary responses to light and near have been reported in a Gypsy family with HMSN-Lom.22 No reports have analysed the pupils in hereditary neuropathy with liability to pressure palsies (HNPP).
In this study, we have analysed the genetic aetiology and the pupil abnormalities in a prospective series of 131 cases and cases presenting between 1998 and 2007 with inherited neuropathy. The patients seen formed five broad groups of inherited neuropathy: FAP, CMT, HNPP, HSAN, and Refsum's disease.
Materials and methods
Patients
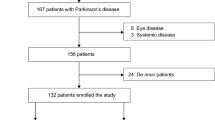
Ethics approval was obtained from the joint medical and ethics committee at The National Hospital for Neurology and Neurosurgery to perform these clinical and genetic studies on hereditary neuropathy. Cases were ascertained through either the genetic peripheral nerve clinic run by one of the authors (MMR), the neurogenetics clinic (HH) (NHNN), and those patients with characterized neuropathies referred for slit lamp microscopy and pupillography. In addition to neurological examination, all patients were assessed for autonomic features in their history and cardiac examination and lying and standing blood pressure calculations were carried out. One hundred and thirty one patients (57 male/74 female patients, aged 7–85 years) were recruited to the study. In the patients studied, the ocular history was reviewed to exclude patients with a blind eye, trauma, iritis, and conditions that could also affect pupil size and function. The medication history was also reviewed to exclude patients on narcotics, anticholinergics, pilocarpine eyedrops, and other drugs that could affect the pupils. The clinical diagnoses, sex distribution, and age range are given in Table 1.
Genetic diagnoses
DNA was extracted from blood samples obtained with informed consent from patients. DNA analysis was carried out in 126 patients; in 5 patients analysis could not be completed. The chromosome 17 duplication and deletion analysis and genetic sequencing of other genes has been described previously, but primers and methods are available on request23 (http://www.molgen.ua.ac.be/CMT).
Pupillography
Pupil diameters and their responses to light and accommodation (near) were recorded with a Whittaker/Applied Science Laboratories infrared television pupillometer as described previously.24 Bilateral recordings were made wherever possible and darkness anisocoria and right–left diameter differences recorded. For clarity and statistical analysis, the remaining measurements for only one eye are presented (right if available) per patient. Reduced pupil diameters in darkness and bilateral Horner's syndrome were taken as indicators of sympathetic dysfunction, whereas reduced light reflexes and/or mydriasis in the light, bilateral pupillotonia, and light–near dissociation as indicators of parasympathetic dysfunction.
Statistics
All measurements were compared with those obtained under identical conditions in 315 healthy subjects (172 male/143 female subjects) aged 16–82 years as described and reported previously.24
All patients were subjected to multiple pupil function tests, seven of which define quantitative abnormality as a value lying outside one 97.5% confidence interval. Within this scenario, in the assessment of individual patients, single quantitative abnormalities have a probability value of 0.150 and they have been ignored. Two abnormalities with a value P=0.012 and single abnormalities lying outside one 99.5% confidence interval (P=0.034) have been included. Bilateral tonic pupils and bilateral Horner's syndrome do not occur in healthy subjects; such abnormalities have been included.
Results
The clinical diagnosis and the pupil abnormalities in the 131 patients are summarized in Table 1. The genetic defects identified along with the individual patient findings are given in detail in Tables 2 and 3. Table 4 shows a comparison of pupil abnormalities in the different groups. Refsum's disease was diagnosed in 11 patients, based on raised phytanic acid. In the remaining 120 cases, 84 had a genetic diagnosis (70%). Overall, at least one significant pupil abnormality was found in 45/131 (34.4%) patients. We discuss only selected results as a great deal of information is given in the tables.
Familial amyloid polyneuropathy
In the FAP group, we have found a high prevalence of pupil abnormality in the 16 patients studied (Tables 1, 2 and 3). Two had bilateral tonic pupils with anisocoria, reduced light reflexes, but normal near responses. Of the other 14 patients, 8 had bilateral Horner's syndrome with redilatation lag (Figure 1). The remaining six had normal pupils. Out of the 10 patients with pupil abnormality, 8 had signs of autonomic dysfunction elsewhere, mainly blood pressure and cardiac abnormalities (Tables 2 and 3).

Dark diameter (graphs on the left), the relationship between age and pupil dark diameter in healthy control subjects (top—normals), all patients with inherited neuropathy included in this study (middle—all patients), and patients with Refsum's disease (bottom—Refsum). Redilatation, relationship between light reflex amplitude, and redilatation time in healthy control subjects (top), all patients with inherited neuropathy (middle), and patients with familial amyloid neuropathy (bottom—FAP).
Charcot Marie Tooth disease
In the CMT group, 12 of the 69 patients had abnormal pupils (Tables 2 and 3). Of the 12 pupil abnormalities, 8 were in the CMT2 group. The difference in occurrence of abnormality between clinical types is statistically significant (χ2=20.09, d.f.=7, P=0.005), with 42.1% of the CMT 2 patients having abnormal pupils. Three patients, all clinically CMT 2 (one in a recessive family), had bilateral Horner's syndrome. All were negative for Chromosome 17 duplication, two were negative for MPZ and Connexin mutations. In one patient, a mutation in the Mitofusin 2 gene was identified, case 13 (MFN2 Arg94Gln). This young female patient had a mild axonal neuropathy along with marked physiological anisocoria, outside the normal range (P<0.01), and the smaller pupil was abnormally small for her age. Unfortunately, no further MFN2 mutations have been identified to compare the pupil abnormalities. Fifty-seven patients had normal pupils. All six HNPP patients studied had normal pupils.
Refsum's disease
In Refsum's disease, 9 patients out of 11 had abnormally small pupils, 7 being below the 0.5% confidence limit (Figure 1). A total of 3/7 showed reduced light reflexes, 2/6 showed exaggerated near responses, and 3/6 showed light–near dissociation. None of the reflexes, although abnormal, was truly tonic. One patient had normal pupils. Tropicamide 0.5%, followed 30 min later by phenylephrine 10% eye drops, was instilled into one eye of 10 of these 11 patients. The final diameter reached was 6.00±0.64 mm (range 2.8–8.4 mm).
Hereditary sensory and autonomic neuropathy
The HSAN patients consisted of 13 (44.8%) out of 29 with abnormal pupils. In patients with the SPTLC1 C133W mutation, three out of four had abnormal pupils, and in the non-HSAN-I cases, there was a much higher incidence of pupil abnormalities. Both patients with HSAN-IV had abnormal pupils; both of these patients had a novel frameshift mutation in exon 15 of the TRKA gene (Table 2). In one patient with the TRKA mutation, pupillometry was impossible but photography revealed miosis below the 0.5% confidence interval of normal. The other had bilateral Horner's syndrome, also with severe miosis. The proband with HSAN-V (homozygous point mutation in exon 8 of the TRKA gene) had tonic pupils as did his father, who was a heterozygous carrier and was clinically normal otherwise.
Discussion
In this series of 131 patients with hereditary neuropathies, we provide an overview of the frequency and type of pupil defects. The neuropathy types were split into five groups, with the Refsum's disease and the autonomic neuropathies (FAP and HSAN) having the greatest frequency of defects. The severity of the autonomic neuropathy in the cases of FAP and those with HSAN was associated with a greater likelihood of having abnormal pupils, but this was not in all cases or all mutation types. This was most consistent in the patients with HSAN-III, -IV, and -V who all had pupil abnormalities and a severe neuropathy with autonomic features. There were cases of HSAN-I, HSAN-II, and FAP with moderate autonomic neuropathy but no pupil abnormalities. This is consistent with previous data on the small group of FAP and Refsum's patients but unreported in HSAN. In the parents available from recessive families with HSAN-II and HSAN-V, abnormal pupils were also recognized, suggesting that they were mild manifesting carriers.
The most frequent FAP group observed was the Ala60 TTR mutation (Irish type) seen in four cases. The main pupil abnormality seen was bilateral Horner's syndrome, reported also in light-chain-associated amyloidosis.25 The two cases with Met30 mutations did not have abnormal pupils, which was unexpected, as a previous study by Ando et al26 reported that 81% of Met30 patients had abnormal pupils early in their diagnosis.
The frequency of pupil abnormalities in CMT and HNPP was low and not associated with severity or mutation type, although one unexpected finding was the frequency of pupil abnormalities in CMT 2, especially as only one of the CMT 2 group with pupil abnormalities had an MPZ mutation. It is well recognized that MPZ mutations are a frequent cause of abnormal pupils in CMT 2 as in one of our cases.20, 23 It is, however, apparent that not all MPZ mutations cause pupil abnormalities, because we found normal pupils in cases with CMT1B and CMT2 and MPZ mutations. The majority of our CMT2 cases were also screened for the Mitofusin 2 gene, a frequent cause of CMT2A (Table 2). Only one case was found to have an MFN2 mutation and she had marked anisocoria. The phenotype spectrum of MFN2 mutations includes axonal neuropathy with optic atrophy, but there are no previous reports of pupil abnormalities.27 To confirm the association of abnormal pupils with MFN2 mutations, further cases need to be identified and examined. The frequency of pupil abnormalities in the CMT 2 group negative for MPZ and MFN2 suggests that the axonal neuropathy present in this form of CMT may in itself be associated with pupil abnormality or the unknown genetic defect(s) lead to abnormal deposits in the iris.
In the group with Refsum's disease, only one patient did not have a pupil abnormality. This case had an extremely strict diet and undetectable phytanic acid levels at the time of pupillography. The main abnormality found in the other patients was miosis.28, 29 The relative failure of the pupils of Refsum's disesase patients to dilate to phenylephrine suggests that in this condition, miosis is not due to an autonomic defect, but it is more likely due to a structural abnormality within the iris. This is consistent with previous pathological and electron microscopy examinations of the irides which have shown high concentrations of the phytanic acid lipid deposits in both sphincter and dilator muscles.30, 31 Any structural abnormality would correlate with the severity of the Refsum's disease and respond to diet restriction if started early enough. There are unfortunately no neuropathological studies on the irides of the other forms of inherited neuropathies in our cohort.
In summary, this is the first study of the pupil abnormalities found in a large group of inherited peripheral neuropathies and provides an overview of the frequency and type of defects seen but analysing this data we could not predict the genetic defect. In the FAP and HSAN (non-HSAN-I) groups, abnormal pupils were associated with a more severe neuropathy, but not in the other inherited neuropathy groups. This series along with the detailed tables will act as an important diagnostic aid in assessing these patients.
References
Skre H . Genetic and clinical aspects of Charcot–Marie–Tooth's disease. Proceedings of the Third International Congress on Muscle Diseases. Excerpta Med Int. Cong. Series, No 334. Amsterdam, Excepta Medica, 1974. 1974.
Zuchner S, Vance JM . Molecular genetics of autosomal-dominant axonal Charcot–Marie–Tooth disease. Neuromolecular Med 2006; 8: 63–74.
Szigeti K, Nelis E, Lupski JR . Molecular diagnostics of Charcot–Marie–Tooth disease and related peripheral neuropathies. Neuromolecular Med 2006; 8: 243–254.
Pareyson D, Scaioli V, Laura M . Clinical and electrophysiological aspects of Charcot–Marie–Tooth disease. Neuromolecular Med 2006; 8: 3–22.
Nicholson G, Myers S . Intermediate forms of Charcot–Marie–Tooth neuropathy: a review. Neuromolecular Med 2006; 8: 123–130.
Kleopa KA, Scherer SS . Molecular genetics of X-linked Charcot–Marie–Tooth disease. Neuromolecular Med 2006; 8: 107–122.
Houlden H, Reilly MM . Molecular genetics of autosomal-dominant demyelinating Charcot–Marie–Tooth disease. Neuromolecular Med 2006; 8: 43–62.
Dubourg O, Azzedine H, Verny C, Durosier G, Birouk N, Gouider R et al. Autosomal-recessive forms of demyelinating Charcot–Marie–Tooth disease. Neuromolecular Med 2006; 8: 75–86.
Dyck P, Chance P, Lebo R, Carney J . Hereditary Motor and Sensory Neuropathies. W.B. Saunders: Philadelphia, 1993.
Lipton L, Tomlinson I . The genetics of FAP and FAP-like syndromes. Fam Cancer 2006; 5: 221–226.
Duke JR, Paton D . Primary familial amyloidosis: ocular manifestations with histopathologic observations. Trans Am Ophthalmol Soc 1965; 63: 146–167.
Wong VG, McFarlin DE . Primary familial amyloidosis. Arch Ophthalmol 1967; 78: 208–213.
Lessell S, Wolf PA, Benson MD, Cohen AS . Scalloped pupils in familial amyloidosis. N Engl J Med 1975; 293: 914–915.
Davies DR, Smith SE . Pupil abnormality in amyloidosis with autonomic neuropathy. J Neurol Neurosurg Psychiatry 1999; 67: 819–822.
Murakami A, Fujiki K, Hasegawa S, Imamura S, Kawano H, Kanai A et al. Transthyretin Ser-44 mutation in a case with vitreous amyloidosis. Am J Ophthalmol 2002; 133: 272–273.
Murakami T, Atsumi T, Maeda S, Tanase S, Ishikawa K, Mita S et al. A novel transthyretin mutation at position 30 (Leu for Val) associated with familial amyloidotic polyneuropathy. Biochem Biophys Res Commun 1992; 187: 397–403.
Refsum S . Heredoataxia hemeralopica polyneuritiformis—ettidligere ikke beskrevet familiaert syndrom? Nord Med 1945; 28: 2682.
Clarke D, Critchley M . Heredopathia atactica polyneuritiformis (Refsum's syndrome). Proc Roy Soc Med 1951; 44: 689–690.
Dyck PJ, Schaid DJ . Genetic heterogeneity in hereditary sensory and autonomic neuropathies: the need for improved ascertainment. Muscle Nerve 2000; 23: 1453–1455.
Kurihara S, Adachi Y, Wada K, Adachi A, Ohama E, Nakashima K . Axonal and demyelinating forms of the MPZ Thr124Met mutation. Acta Neurol Scand 2003; 108: 157–160.
Reilly M, Greenwood RJ . Disorders of the peripheral nerves. In: Greenwood RJ, Barnes MP, McMillan TM, Ward CD (eds). Handbook of Neurological Rehabilitation. Psychology Press, Taylor and Francis Group: Hove and New York, 2003, vol 1, pp 725.
Leonardis L, Zidar J, Popovic M, Timmerman V, Löfgren A, Van Broeckhoven C et al. Hereditary motor and sensory neuropathy associated with auditory neuropathy in a Gypsy family. Pflugers Arch 2000; 439: R208–R210.
Reilly MM . Sorting out the inherited neuropathies. Pract Neurol 2007; 7: 93–105.
Bremner F, Smith S . Pupil findings in a consecutive series of 150 patients with generalised autonomic neuropathy. J Neurol Neurosurg Psychiatry 2006; 77: 1163–1168.
Bremner FD, Smith SE . Pupil abnormalities in selected autonomic neuropathies. J Neuroophthalmol 2006; 26: 209–219.
Ando E, Ando Y, Okamura R, Uchino M, Ando M, Negi A . Ocular manifestations of familial amyloidotic polyneuropathy type I: long-term follow up. Br J Ophthalmol 1997; 81: 295–298.
Zuchner S, De Jonghe P, Jordanova A, Claeys KG, Guergueltcheva V, Cherninkova S et al. Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann Neurol 2006; 59: 276–281.
Folz SJ, Trobe JD . The peroxisome and the eye. Surv Ophthalmol 1991; 35: 353–368.
Claridge KG, Gibberd FB, Sidey MC . Refsum disease: the presentation and ophthalmic aspects of Refsum disease in a series of 23 patients. Eye 1992; 6 (Part 4): 371–375.
Toussaint D, Danis P . An ocular pathologic study of Refsum's syndrome. Am J Ophthalmol 1971; 72: 342–347.
Dick AD, Jagger J, McCartney AC . Refsum's disease: electron microscopy of an iris biopsy. Br J Ophthalmol 1990; 74: 370–372.
Acknowledgements
We are grateful to the patients, without whom the study would not have been possible. We also thank the MRC, HH holds an MRC clinician scientist fellowship. Professor Smith died in December 2007 during the final preparation of this manuscript. He made an enormous contribution to this paper and to the investigation of pupil abnormalities throughout his life.
Author information
Authors and Affiliations
Corresponding author
Additional information
There are no conflicts of interest and this work has not been previously presented or published
Professor Smith died in December 2007
Supplementary Information accompanies the paper on Eye website (http://www.nature.com/eye)
Supplementary information
Rights and permissions
About this article
Cite this article
Houlden, H., Reilly, M. & Smith, S. Pupil abnormalities in 131 cases of genetically defined inherited peripheral neuropathy. Eye 23, 966–974 (2009). https://doi.org/10.1038/eye.2008.221
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2008.221
Keywords
This article is cited by
-
Ophthalmic manifestations of inherited neurodegenerative disorders
Nature Reviews Neurology (2014)
