
Abstract
Purpose
To describe ocular abnormalities in patients with Friedreich ataxia (FRDA).
Methods
Patients diagnosed with FRDA by genetic analysis were invited to participate in a prospective cohort. The patients included underwent an extensive ophthalmologic examination, including low-contrast Sloan letter charts test and retinal nerve fiber layer (RNFL) thickness analysis by optical coherence tomography (OCT).
Results
Twenty-three patients agreed to participate. In all, 19 patients (83%) had a visual acuity of at least 0.8 in both eyes. Fundus examination showed diffuse optic nerve pallor in four patients. However, OCT showed a decreased mean peripapillary RNFL thickness in all but three adult cases and one teenager. The RNFL thickness was found to have a positive correlation with visual acuity (P=0.001) and contrast sensitivity (P=0.001) and a negative correlation with time elapsed from diagnosis (P=0.001).
Conclusions
OCT and low contrast test sensitivity show that the visual pathway is affected in FRDA. However, in most patients there is no significant visual impairment. In a small proportion of patients visual acuity declines with disease progression. This study provides a better understanding of the ophthalmic features of FRDA.
Similar content being viewed by others

Introduction
Friedreich ataxia (FRDA) is the most frequent form of hereditary ataxia among Caucasians, affecting about 1 in 30 000 individuals in Western Europe. It is a neurodegenerative disease, which affects the central and peripheral nervous systems and has an autosomal recessive pattern of inheritance.1 The disease is characterized by progressive limb and gait ataxia, leg weakness, dysarthria, hypertrophic cardiomiopathy, hypoacusia, and an increased incidence of diabetes mellitus.1, 2, 3 Symptoms usually begin before the second decade of life.
Almost all individuals with FRDA are reported to be homozygous for a GAA triplet repeat expansion in intron 1 of the frataxin gene and only 2–5% of them are heterozygous. This expansion results in an abnormal influx of iron into the mitochondria, which increases the susceptibility of the nervous system, including the visual pathway, to oxidative stress.1, 2, 4
FDRA is usually an insidious disease, with a slow decline in muscular function so that most patients lose their ability to walk, stand, or sit without support within 10–15 years of onset.1 The rate of progression is neither constant nor linear, and varies among individuals and at the different stages of the disease.1, 2
Ocular motor abnormalities are the best-characterized signs of damage to the visual system in patients with FRDA. They reflect the disruption of the brainstem–cerebellar circuit and include fixation instability, saccadic dysmetria, disrupted pursuit, and vestibular abnormalities. The most common manifestation is fixation instability with frequent square-wave jerks.1, 2, 3 To date, visual ophthalmic manifestations have been described in up to 30% of patients, including optic neuropathy and, less commonly, a retinitis pigmentosa-like syndrome.
Several tools are employed to evaluate progression in FDRA and most of them are based on neurological scales. However, none has been proven to be superior for patient evaluation in clinical trials.
This study aimed to characterize the ocular abnormalities in FDRA and to correlate our findings with neurological disability and disease duration.
Materials and methods
Patients diagnosed with FDRA (genetically confirmed) were identified from our hospital's records and invited to participate in this observational case series. They were recruited from La Paz University Hospital (Madrid, Spain), a national reference center for ataxias. Informed consent was obtained from every patient and the study was approved by the local ethics committee.
A detailed medical history was obtained. Disability was quantified according to two scales, the FRDA Rating Scale (FARS) and the International Cooperative Ataxia Rating Scale (ICARS). The former comprises three subscales, a general score for ataxia, a score for activities of daily living, and a neurological examination. The scores can be added to make a total score ranging from 0 to 159; a higher score indicates a greater level of disability. The ICARS test is a 100-point scale divided into the following sections: postural and stance disturbances (maximum score: 34); limb movement disturbances (52); speech (8); and oculomotor dysfunction (6). Each side of the body is assessed separately and the sum of scores for both sides is added to obtain the final score. A higher score also indicates more disability.
Patients were classified according to the number of GAA-triplet repetitions into three groups: <500 repetitions, between 500 and 700, and >700 (as provided by the genetic laboratory). For this classification, the allele with the lowest number of repetitions (less-affected allele) was selected.
Patients underwent an extensive ophthalmic examination, including monocular and binocular visual acuity and binocular low contrast sensitivity. We employed the high-contrast ETDRS chart at 3.2 m and two low-contrast (2.5 and 1.25%) Sloan charts (LCSLC, Precision Vision, LaSalle, IL, USA) at 2 m. Contrast acuity was expressed as a score out of 70, calculated depending on the number of letters correctly identified in each chart. Monocular high-contrast visual acuity was noted in decimal and logarithm of the minimum angle of resolution (logMAR) scales, both provided by the ETDRS chart. LogMAR scale data were used for statistical analysis.
We also performed anterior and posterior pole biomicroscopy, internal and external ocular motor examinations, funduscopy, and optical coherence tomography (OCT). OCT scanning was performed with the Stratus OCT (Carl Zeiss Meditec, Dublin, CA, USA) after pharmacological mydriasis using internal fixation whenever possible. Fast retinal nerve fiber layer (RNFL) and fast macular thickness acquisition protocols were used. Peripapillary RNFL thickness values were obtained with the built-in software RNFL Thickness Average analysis protocol. A color-coded graph displays the RNFL measurements and compares them with age-matched data. For the purposes of statistical analysis, RNFL thickness values that were colored yellow or red by the normative database were considered reduced. Central macular thickness, an area 1 mm in diameter and centered on the fovea, was obtained using the retinal thickness/volume analysis protocol.
Results
Twenty-three patients with genetically confirmed FDRA were recruited. All of them had >300 GAA-triplet repetitions. In all, 14 patients were women (61%) and 9 patients were men (39%), and their ages ranged between 9 and 34 years at the time of the ophthalmic evaluation. Table 1 summarizes demographic and disease-related data. Only one patient reported symptomatic visual impairment; he presented with unilateral amblyopia due to childhood esotropia.
Visual acuity ranged between 0.1 and 1 in the right eye (OD) and between 0.4 and 1 in the left eye (OS). Table 2 provides more details of visual acuity and low-contrast test data.
External ocular movements were normal in 15 patients (65.2%), 1 patient had childhood esotropia (4.3%), and spontaneous horizontal nystagmus was recorded in 7 patients (30.4%). During anterior biomicroscopy, traumatic corneal leucomas were detected in two patients; intraocular pressure was <18 mm Hg in all cases.
Fundus examination revealed no pigmentary or vascular abnormalities; however, two cases had diffuse optic disc pallor in both eyes (OU). Another patient had localized temporal optic disc pallor in OU.
Macular morphology and thickness were normal in all patients (Table 3). Peripapillary RNFL thickness acquisition was possible in 16 OD and 17 OS of the adult patients. Mean RNFL thickness was within normal values in 4 right eyes (25%) and was considered reduced in the remaining 12 right eyes (4 (25%) under percentile 5% and in 8 eyes (50%) under percentile 1%). In all, 5 left eyes (21.7%) had an average RNFL thickness within normal parameters, 6 left eyes (26.1%) under percentile 5%, and 6 left eyes (26.1%) under percentile 1%. Therefore, only 9 eyes were considered normal and 24 showed some degree of atrophy by OCT criteria. Only three adults showed a mean RNFL thickness within normal parameters in both eyes.
Mean RNFL thickness values obtained in the four patients who were underage ranged between 80.11 and 108.44 μm. In a study performed to obtain normal values for OCT in a pediatric population, the median RNFL thickness was 108 μm and the percentile 5% cutoff was 92 μm.5 In all, 5 out of 8 eyes of our patients were below this percentile.
Mean RNFL thickness was significantly correlated with all binocular test scores, with time elapsed from diagnosis and with ICARS score (Table 4). Low contrast sensitivity was also significantly correlated with ICARS score (R: 0.579; P=0.004 for the 2.5% chart and R: 0.74 P=0.001 for the 1.25% chart). No significant correlations were found for the FARS scale. As the number of GAA-triplet repetitions increased, mean RNFL thickness tended to decrease; however, differences between groups were not statistically significant (Table 5).
Discussion
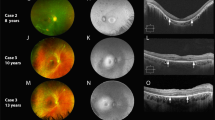
Friedreich ataxia usually presents before the age of 25 and does not have gender predilection. We have studied a relatively young population of FRDA patients with a slight female dominance. Previous reports have focused on ocular movement disorders;6, 7, 8, 9, 10, 11 instead, we focused on other ocular abnormalities. We found that even though only one patient complained of visual loss, contrast sensitivity (the ability to distinguish adjacent areas of differing luminance) was abnormal in most cases. OCT is a quick test, easily performed, useful in neurologically disabled subjects like FRDA patients, for whom functional tests such as visual fields can be discouraging. OCT provides information that years ago could only be subjectively obtained by funduscopy and is sensitive to decreased RNFL thickness when visual impairment is not clinically apparent.1, 4, 12 Indeed, in our study, 24 out of 33 adult eyes (73%) had a decreased RNFL thickness compatible with different degrees of optic atrophy, whereas only 3 eyes seemed to be pale on fundoscopy (Figure 1). Three out of four underage cases also showed decreased mean RNFL thickness. These results suggest that FDRA is associated with a frequent, subclinical optic neuropathy.

Two patients diagnosed with FRDA in different stages of the disease. Even in the most severe case, the thickness of the temporal quadrant is not decreased.
Mitochondrial diseases have a wide spectrum of clinical manifestations. Leber's hereditary optic neuropathy (LHON) and dominant optic atrophy (DOA) produce an optic neuropathy that is characterized by papillomacular bundle damage, with initial centrocecal scotomas and visual acuity loss.4, 13 It has been suggested that in FDRA, reduced frataxin expression leads to decreased intracytoplasmatic iron levels, shifting the redox state out of its optimum levels. This predisposes the retinal ganglion cells to oxidative damage. As the mechanisms for managing this damage are gradually overcome, retinal ganglion cell death takes place. Thus, optic neuropathy would develop with age.2 Our results support this hypothesis, as we found a negative correlation between disease duration and RNFL thickness. Fortuna et al1 found a correlation between age at onset and RNFL thickness (P=0.007), but not between disease duration and RNFL thickness (P=0.068). The difference between these results and ours might be explained by the different study populations: Fortuna et al included FRDA patients with a mean age of 32 years (range 24–40), while the age of our patients ranged between 9 and 34. We believe this wider age range increased the possibilities of detecting a relationship between disease duration and RNFL thickness. Previous studies have established that visual impairment in FRDA is related to the number of GAA repeats; however, we cannot confirm these results. The exact number of repetitions in each case was not available to assess this correlation, and although RNFL thickness tended to decrease as the number of GAA repeats increased, differences were not statistically significant.
A remarkable feature of optic neuropathy in FRDA, as opposed to LHON and DOA, is that it is not clinically apparent despite its frequency. Visual acuity is not usually affected, as occurred in our population, probably because the papillomacular bundle is often preserved.1, 2, 4 In the present study, macular thickness and the RNFL of the temporal quadrant on OCT, which represent the papillomacular bundle, were normal (Figure 1). The reason why the papillomacular bundle is preserved in this disease although it is the one initially damaged in other mitochondrial diseases is unclear. Further investigations on the exact pathophysiological mechanism of each mitochondrial disease might explain the differences in the pattern, timing, and rate of progression of optic neuropathy.
In order to detect mild optic nerve disease, electrophysiological tests, contrast sensitivity, perimetry, and OCT examination are needed.1, 2, 14 Contrast sensitivity has been described to be a more sensitive marker of optic nerve damage than visual acuity,14 as well as a better marker for disease progression.12, 14, 15, 16 In our study, only 18% of patients had optic nerve abnormalities on funduscopy; however, almost all patients had low contrast sensitivity. We also found that contrast sensitivity is correlated with RNFL thickness, making it a potentially useful test for monitoring response to treatment in clinical trials. Therefore, contrast sensitivity should be the functional test to assess progression in FRDA patients.
The FARS is the most widely employed scale to evaluate neurological disability in this disease. The ICARS was developed to evaluate the effect of medical treatment in ataxia. Both have been validated and show good inter-rater reliability.3, 16, 17, 18 Mean RNFL thickness was significantly correlated with the ICARS score (Table 4); however, no significant correlation was found for the FARS scale. This may be due to the different items scored in each scale. The ICARS scale evaluates two items related to visual function: postural and stance disturbances, and oculomotor dysfunction, whereas none of the items scored by the FARS scale are related to visual function.19 Fortuna et al1 found positive correlations between scores obtained in the ICARS test and disease duration, and mean RNFL thickness. However, they did not find a significant correlation with contrast sensitivity as we did.
The search for a valid tool to evaluate disease progression for FRDA is still ongoing.16, 20, 21 Mild optic neuropathy seems to be frequent in patients with FDRA and to be correlated with disease duration. Both contrast sensitivity and OCT are quick, easily performed, non-invasive techniques that are able to detect and quantify early optic nerve damage. They should be routinely performed to evaluate progression and response to treatment in FRDA patients. Longitudinal clinical trials are needed in order to demonstrate if optic neuropathy progression is really correlated with the number of GAA repeats and with systemic disease progression in FRDA.

References
Fortuna F, Barboni P, Liguori R, Valentino ML, Savini G, Gellera C et al. Visual system involvement in patients with Friedreich's ataxia. Brain 2009; 132: 116–123.
Alldredge CD, Schlieve CR, Miller NR, Levin LA . Pathophysiology of the optic neuropathy associated with Friedreich ataxia. Arch Ophthalmol 2003; 121: 1582–1585.
Fahey MC, Cremer PD, Aw ST, Millist L, Todd MJ, White OB et al. Vestibular, saccadic and fixation abnormalities in genetically confirmed Friedreich ataxia. Brain 2008; 131: 1035–1045.
Carelli V, La MC, Valentino ML, Barboni P, Ross-Cisneros FN, Sadun AA . Retinal ganglion cell neurodegeneration in mitochondrial inherited disorders. Biochim Biophys Acta 2009; 1787: 518–528.
El-Dairi MA, Asrani SG, Enyedi LB, Freedman SF . Optical coherence tomography in the eyes of normal children. Arch Ophthalmol 2009; 127: 50–58.
Dale RT, Kirby AW, Jampel RS . Square wave jerks in Friedreich's ataxia. Am J Ophthalmol 1978; 85: 400–406.
Furman JM, Perlman S, Baloh RW . Eye movements in Friedreich's ataxia. Arch Neurol 1983; 40: 343–346.
Hocking DR, Fielding J, Corben LA, Cremer PD, Millist L, White OB et al. Ocular motor fixation deficits in Friedreich ataxia. Cerebellum 2010; 9: 411–418.
Kirkham TH, Guitton D, Katsarkas A, Kline LB, Andermann E . Oculomotor abnormalities in Friedreich's ataxia. Can J Neurol Sci 1979; 6: 167–172.
Monday L, Lesperance J, Lemieux B, Saint-Vincent H . Follow-up study of electronystagmographic findings in Friedreich's ataxia patients and evaluation of their relatives. Can J Neurol Sci 1984; 11: 570–573.
Spieker S, Schulz JB, Petersen D, Fetter M, Klockgether T, Dichgans J . Fixation instability and oculomotor abnormalities in Friedreich's ataxia. J Neurol 1995; 242: 517–521.
Fisher JB, Jacobs DA, Markowitz CE, Galetta SL, Volpe NJ, Nano-Schiavi ML et al. Relation of visual function to retinal nerve fiber layer thickness in multiple sclerosis. Ophthalmology 2006; 113: 324–332.
Koutnikova H, Campuzano V, Foury F, Dollé P, Cazzalini O, Koenig M . Studies of human, mouse and yeast homologues indicate a mitochondrial function for frataxin. Nat Genet 1997; 16: 345–351.
Balcer LJ . Clinical outcome measures for research in multiple sclerosis. J Neuroophthalmol 2001; 21: 296–301.
Arditi A . Improving the design of the letter contrast sensitivity test. Invest Ophthalmol Vis Sci 2005; 46: 2225–2229.
Lynch DR, Farmer JM, Tsou AY, Perlman S, Subramony SH, Gomez CM et al. Measuring Friedreich ataxia: complementary features of examination and performance measures. Neurology 2006; 66: 1711–1716.
Delatycki MB . Evaluating the progression of Friedreich ataxia and its treatment. J Neurol 2009; 256 (Suppl 1): 36–41.
Friedman LS, Farmer JM, Perlman S, Wilmot G, Gomez CM, Bushara KO et al. Measuring the rate of progression in Friedreich ataxia: implications for clinical trial design. Mov Disord 2010; 25: 426–432.
Burk K, Malzig U, Wolf S, Heck S, Dimitriadis K, Schmitz-Hübsch T et al. Comparison of three clinical rating scales in Friedreich ataxia (FRDA). Mov Disord 2009; 24: 1779–1784.
Fahey MC, Corben L, Collins V, Churchyard AJ, Delatycki MB . How is disease progress in Friedreich's ataxia best measured? A study of four rating scales. J Neurol Neurosurg Psychiatry 2007; 78: 411–413.
Lynch DR, Farmer JM, Rochestie D, Balcer LJ . Contrast letter acuity as a measure of visual dysfunction in patients with Friedreich ataxia. J Neuroophthalmol 2002; 22: 270–274.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Noval, S., Contreras, I., Sanz-Gallego, I. et al. Ophthalmic features of Friedreich ataxia. Eye 26, 315–320 (2012). https://doi.org/10.1038/eye.2011.291
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2011.291
Keywords
This article is cited by
-
Retinale OCT-Biomarker und neurodegenerative Erkrankungen des zentralen Nervensystems jenseits der Alzheimer-Krankheit
Die Ophthalmologie (2024)
-
Patient-derived iPSC models of Friedreich ataxia: a new frontier for understanding disease mechanisms and therapeutic application
Translational Neurodegeneration (2023)
-
Memantin bei Optikusatrophie in Friedreich-Ataxie
Der Ophthalmologe (2016)
-
Ophthalmic manifestations of inherited neurodegenerative disorders
Nature Reviews Neurology (2014)
