
Abstract
The Brugada syndrome is characterized by unique ‘coved-type’ ST-segment elevation in the right precordial leads of electrocardiogram and ventricular fibrillation, and is responsible for 4 to 12% of sudden cardiac death in the general population. The frequency is higher in Southeast Asia including Japan compared with Western countries. Brugada syndrome is an inherited disease usually transmitted in an autosomal-dominant manner, and incomplete penetrance is frequently seen within affected families. To date, 20 genes have been associated with Brugada syndrome, but pathogenic mutations in the genes are identified in only about 30% of patients. The genetic background includes mutations in genes encoding sodium channel, calcium channels and potassium channels, as well as proteins affecting ion channels. Mutations in SCN5A, encoding the cardiac predominant sodium channel α-subunit, account for 20 to 30% of patients with Brugada syndrome and mutations in other genes only account for about 5% of patients. Furthermore, a recent genome-wide association study has identified new loci associated with the susceptibility of Brugada syndrome.
Similar content being viewed by others
Clinical characteristics
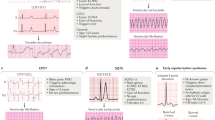
The Brugada syndrome is characterized by unique ‘coved-type’ ST-segment elevation in the right precordial leads (V1–V3) of the 12-lead electrocardiogram and episodes of ventricular fibrillation and sudden cardiac death (Figure 1).1 It is responsible for 4 to 12% of sudden cardiac death in the general population and about 20% of sudden cardiac death in patients with structurally normal hearts.2 Brugada syndrome is more common in Southeast Asia including Japan, with the estimated prevalence of 12/10 000 individuals, compared with western countries, with the estimated prevalence of 1 to 5/10 000 individuals.3 The age at diagnosis is around 40 years and 70 to 80% of patients are men.4, 5, 6, 7 A family history of unexplained sudden death is present in 20 to 30% of patients with Brugada syndrome.4, 5

Electrocardiogram showing typical coved-type ST-segment elevation in lead V1 and V2 in a healthy individual and a patient with Brugada syndrome.
Arrhythmia events resulted from ventricular fibrillation mainly occur during sleep, at rest and after meal.8 The annual incidence of arrhythmia recurrences in survivors of ventricular fibrillation is 7 to 8%.4, 5 In addition to the susceptibility to ventricular fibrillation, atrial fibrillation occurs in 10 to 20% of patients.9 Conduction abnormalities at the His-Purkinje system and the right ventricular outflow tract are sometimes evident.
The characteristic ‘coved-type’ ST-segment elevation is dynamic, and the amplitude and the existence of ST-segment elevation varies. ST-segment elevation augments after pause and during bradycardia, and sometimes disappears. ST-segment elevation becomes most prominent just before the development of ventricular fibrillation, supporting the arrhythmogenicity of ST-segment elevation.1 Because the electrocardiographic changes can occasionally disappear, sodium channel blockers are used to provoke the diagnostic Brugada electrocardiographic pattern in patients with suspected disease.
Genetic basis
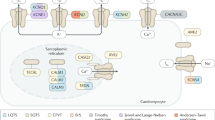
Similar to other arrhythmia syndromes such as long QT syndrome and catecholaminergic polymorphic ventricular tachycardia, Brugada syndrome is an inherited disease usually transmitted in an autosomal-dominant manner.10 Incomplete penetrance is frequently seen within affected families, and up to 60% of patients are sporadic.11 To date, 20 genes have been associated with Brugada syndrome (Table 1). The genetic background includes mutations in genes encoding sodium channel, calcium channels and potassium channels, as well as proteins affecting ion channels. Although there have been a number of causative genes identified, only about 30% of patients are identified to have pathogenic mutations. Mutations in SCN5A, encoding the cardiac predominant pore-forming sodium channel α-subunit, account for 20 to 30% of patients with Brugada syndrome and mutations in other genes only account for about 5% of patients.3, 12, 13
Sodium channel genes
Sodium channel is composed of single pore-forming α-subunit and accessory proteins such as β-subunits that regulate channel function. Mutations in SCN5A are the most frequent genotype of Brugada syndrome and account for about 80% of genotype-positive patients.3, 12 Other genes including sodium channel β-subunit genes (SCN1B,14 SCN2B,15 SCN3B16), GPD1L,17 MOG1,18 SLMAP19 and PKP220 encoding proteins that modify SCN5A channel (Nav1.5) function are also causative genes of Brugada syndrome.
Functional studies mainly using heterologous expression systems demonstrate that loss-of-sodium channel dysfunction by mutations in sodium channel genes and sodium channel-associated genes is the main mechanism of Brugada syndrome.10, 21, 22 These pathogenic mutations usually result in either or both of (1) decreased expression of the sodium channel protein because of failure to traffic to the cell membrane or expression of non-functional channels or (2) decreased inward sodium current by altered channel gating because of delayed activation, earlier inactivation, faster inactivation, enhanced slow inactivation and delayed recovery from inactivation.
Dominant-negative effects caused by mutations in ion channel genes are frequently found in ion channels formed by multiple α-subunits, such as potassium channel genes that account for long QT syndrome.23 Although cardiac sodium channel α-subunit is not known to oligomerize, α-subunit interaction of sodium channels and dominant-negative effects have been reported.24 Two N-terminal mutant channels have caused dominant-negative effects on wild-type channels, and mutations in SCN5A with dominant-negative effects may result in severe phenotypes.
Mutations in SCN10A, encoding the sodium channel α-subunit mainly expressed in the sensory neurons of dorsal root ganglia and weakly expressed in the hearts, has recently been associated with Brugada syndrome.25, 26 Although mutations in SCN10A caused decreased sodium currents, SCN10A channel dysfunction may not solely cause Brugada syndrome because of very low levels of SCN10A expression in cardiomyocytes.25 Recent studies have provided the possible mechanisms by which mutations in SCN10A cause Brugada syndrome.26, 27 In a previous study using chromatin immunoprecipitation sequencing analysis, SCN10A locus regulates SCN5A expression.27 In heterologous expression systems, both expressing SCN5A and SCN10A, wild-type SCN10A increases sodium currents, but SCN10A mutations reduce sodium currents.26
Calcium channel genes
Similar to sodium channel, calcium channel is composed of single pore-forming α-subunit and accessory proteins. Mutations in the α1- (CACNA1C), β2- (CACNB2) and α2δ- (CACNA2D1) subunit genes of cardiac L-type channels are account for 2 to 4% of patients with Brugada syndrome.13, 28, 29 L-type calcium channel regulates action potential dome and thus calcium channel dysfunction can result in abnormal action potential (QT interval) duration. Actually, L-type calcium channel gene is one of the causative genes of long QT syndrome. Mutations in L-type calcium channel genes were initially identified in patients with overlapping phenotypes of Brugada syndrome and short QT syndrome,28 and then were identified in those with Brugada syndrome and normal QT interval.13, 29 The frequency of mutations in L-type calcium channel genes is high in patients with overlapping phenotypes of Brugada syndrome and short QT syndrome, but is rare in those with Brugada syndrome without short QT interval. Functional analysis data of mutant L-type calcium channels is very limited. Mutations identified in patients with overlapping phenotypes of Brugada syndrome and short QT syndrome and those in patients with Brugada syndrome without short QT interval similarly cause decreased calcium current.28, 30
In addition to mutations resulting in substitution of amino acids, a synonymous mutation in CACNA1C, which does not change amino acids, has been associated with Brugada syndrome.31 mRNA levels of CACNA1C are decreased in mutation carriers compared with those in a non-carrier within a family affected by Brugada syndrome.
Potassium channel genes
Seven potassium channel genes and one gene encoding semaphorin 3A, which blocks potassium channel, are causative genes of Brugada syndrome.32, 33, 34, 35, 36, 37, 38, 39, 40 Although there are multiple causative genes of Brugada syndrome affecting potassium channel function, the frequency of mutations in these genes is very rare.
Loss of the action potential dome is one of the proposed mechanisms for Brugada syndrome. Transient outward potassium channel, which creates action potential notch, is composed of an α-subunit encoded by KCND3 and a β-subunit encoded by KCNE3. Mutations in KCND3 and KCNE3 identified in patients with Brugada syndrome result in the increase of transient outward potassium current, possibly resulting in deep action potential notch and loss of the action potential dome.32, 37 Furthermore, KCNE5 encoding another potassium channel β-subunit is a causative gene of Brugada syndrome and mutations in KCNE5 also result in increase of transient outward potassium current and loss of the action potential dome.38 Semaphorin 3A inhibits transient outward potassium current. Mutations in SEMA3A have recently been identified in patients with Brugada syndrome and mutant semaphorin fails to inhibit transient outward potassium current, resulting in the increase of transient outward potassium current.39
ATP-sensitive potassium channel is composed of an α-subunit encoded by KCNJ8/KCNJ11 and a β-subunit encoded by ABCC9. Gain-of-function mutations in KCNJ8 and ABCC9 have been identified in patients with Brugada syndrome, but the precise mechanism is unknown.40, 41
KCNH2 encoding the rapid component of delayed rectifier potassium channel, which is an important determinant of action potential repolarization, and thus is one of the major causative genes of long QT syndrome and short QT syndrome. Gain-of-function mutations in KCNH2 have initially been identified in patients with Brugada syndrome and normal QT interval, and then have been identified in those with Brugada syndrome and short QT interval.33, 34 Mutant channels show increase of potassium current and change of voltage dependence in functional analysis, and produce loss of action potential dome in simulation model.
Genetic modifiers
In addition to rare variants, common variants have also been associated with Brugada syndrome. A common polymorphism in SCN5A, H558R, on a different allele to a pathogenic mutation in SCN5A has been shown to restore trafficking defect of mutant sodium channels resulting in normalized sodium current, and individuals carrying both the polymorphism and the mutation do not have Brugada phenotype in a family affected by the disease.42
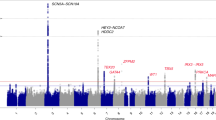
A genome-wide association study is used to examine the association of many common genetic variants selected from the whole genome with a trait. Genome-wide association study typically focuses on associations of single-nucleotide polymorphisms with major traits. In the cardiovascular field, genome-wide association study has been performed for electrocardiographic indices such as PR interval and QT interval in electrocardiogram and for common diseases such as hypertension and atrial fibrillation. However, a recent genome-wide association study for Brugada syndrome has succeeded to identify two common genetic variants in SCN5A-SCN10A and HEY2 associated with the rare disease.43 SCN5A is the major causative gene and SCN10A is also one of the causative genes in Brugada syndrome as shown above. HEY2 encodes a transcriptional repressor in the cardiovascular system, and has been shown to regulate cardiac electrophysiology. However, the mechanism by which variants in HEY2 affects the disease susceptibility is unclear.
References
Brugada, P. & Brugada, J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J. Am. Coll. Cardiol. 20, 1391–1396 (1992).
Juang, J. M. & Huang, S. K. Brugada syndrome—an under-recognized electrical disease in patients with sudden cardiac death. Cardiology 101, 157–169 (2004).
Berne, P. & Brugada, J. Brugada syndrome 2012. Circ. J. 76, 1563–1571 (2012).
Probst, V., Veltmann, C., Eckardt, L., Meregalli, P. G., Gaita, F., Tan, H. L. et al. Long-term prognosis of patients diagnosed with Brugada syndrome: results from the FINGER Brugada Syndrome Registry. Circulation 121, 635–643 (2010).
Kamakura, S., Ohe, T., Nakazawa, K., Aizawa, Y., Shimizu, A., Horie, M. et al. Long-term prognosis of probands with Brugada-pattern ST elevation in V1–V3 leads. Circ. Arrhythm. Electrophysiol. 2, 495–503 (2009).
Haissaguerre, M., Sacher, F., Nogami, A., Komiya, N., Bernard, A., Probst, V. et al. Characteristics of recurrent ventricular fibrillation associated with inferolateral early repolarization role of drug therapy. J. Am. Coll. Cardiol. 53, 612–619 (2009).
Watanabe, H., Nogami, A., Ohkubo, K., Kawata, H., Hayashi, Y., Ishikawa, T. et al. Clinical characteristics and risk of arrhythmia recurrences in patients with idiopathic ventricular fibrillation associated with early repolarization. Int. J. Cardiol. 159, 238–240 (2012).
Shimizu, W. The Brugada syndrome—an update. Intern. Med. (Tokyo, Japan) 44, 1224–1231 (2005).
Antzelevitch, C., Brugada, P., Borggrefe, M., Brugada, J., Brugada, R., Corrado, D. et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 111, 659–670 (2005).
Antzelevitch, C. Genetic, molecular and cellular mechanisms underlying the J wave syndromes. Circ. J. 76, 1054–1065 (2012).
Campuzano, O., Brugada, R. & Iglesias, A. Genetics of Brugada syndrome. Curr. Opin. Cardiol 25, 210–215 (2010).
Chen, Q., Kirsch, G. E., Zhang, D., Brugada, R., Brugada, J., Brugada, P. et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 392, 293–296 (1998).
Crotti, L., Marcou, C. A., Tester, D. J., Castelletti, S., Giudicessi, J. R., Torchio, M. et al. Spectrum and prevalence of mutations involving BrS1- through BrS12-susceptibility genes in a cohort of unrelated patients referred for Brugada syndrome genetic testing: implications for genetic testing. J. Am. Coll. Cardiol. 60, 1410–1418 (2012).
Watanabe, H., Koopmann, T. T., Le Scouarnec, S., Yang, T., Ingram, C. R., Schott, J. J. et al. Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J. Clin. Invest. 118, 2260–2268 (2008).
Riuro, H., Beltran-Alvarez, P., Tarradas, A., Selga, E., Campuzano, O., Verges, M. et al. A missense mutation in the sodium channel beta2 subunit reveals SCN2B as a new candidate gene for Brugada syndrome. Hum. Mutat. 34, 961–966 (2013).
Hu, D., Barajas-Martinez, H., Burashnikov, E., Springer, M., Wu, Y., Varro, A. et al. A mutation in the beta 3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ. Cardiovasc. Genet. 2, 270–278 (2009).
Van Norstrand, D. W., Valdivia, C. R., Tester, D. J., Ueda, K., London, B., Makielski, J. C. et al. Molecular and functional characterization of novel glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) mutations in sudden infant death syndrome. Circulation 116, 2253–2259 (2007).
Kattygnarath, D., Maugenre, S., Neyroud, N., Balse, E., Ichai, C., Denjoy, I. et al. MOG1: a new susceptibility gene for Brugada syndrome. Circ Cardiovasc Genet. 4, 261–268 (2011).
Ishikawa, T., Sato, A., Marcou, C. A., Tester, D. J., Ackerman, M. J., Crotti, L. et al. A novel disease gene for Brugada syndrome: sarcolemmal membrane-associated protein gene mutations impair intracellular trafficking of hNav1.5. Circ. Arrhythm. Electrophysiol. 5, 1098–1107 (2012).
Cerrone, M., Lin, X., Zhang, M., Agullo-Pascual, E., Pfenniger, A., Chkourko Gusky, H. et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation 129, 1092–1103 (2014).
Veerakul, G. & Nademanee, K. Brugada syndrome. Circ. J. 76, 2713–2722 (2012).
Li, A., Saba, M. M. & Behr, E. R. Genetic biomarkers in Brugada syndrome. Biomarkers Med. 7, 535–546 (2013).
Schwartz, P. J., Crotti, L. & Insolia, R. Long-QT syndrome: from genetics to management. Circ. Arrhythm. Electrophysiol. 5, 868–877 (2012).
Clatot, J., Ziyadeh-Isleem, A., Maugenre, S., Denjoy, I., Liu, H., Dilanian, G. et al. Dominant-negative effect of SCN5A N-terminal mutations through the interaction of Na(v)1.5 alpha-subunits. Cardiovasc. Res. 96, 53–63 (2012).
Behr, E. R., Savio-Galimberti, E., Barc, J., Holst, A. G., Petropoulou, E., Prins, B. P. et al. Role of common and rare variants in SCN10A: results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc. Res. 106, 520–529 (2015).
Hu, D., Barajas-Martinez, H., Pfeiffer, R., Dezi, F., Pfeiffer, J., Buch, T. et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J. Am. Coll. Cardiol. 64, 66–79 (2014).
van den Boogaard, M., Smemo, S., Burnicka-Turek, O., Arnolds, D. E., van de Werken, H. J., Klous, P. et al. A common genetic variant within SCN10A modulates cardiac SCN5A expression. J. Clin. Invest. 124, 1844–1852 (2014).
Antzelevitch, C., Pollevick, G. D., Cordeiro, J. M., Casis, O., Sanguinetti, M. C., Aizawa, Y. et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 15, 442–449 (2007).
Fukuyama, M., Ohno, S., Wang, Q., Kimura, H., Makiyama, T., Itoh, H. et al. L-type calcium channel mutations in Japanese patients with inherited arrhythmias. Circ. J. 77, 1799–1806 (2013).
Burashnikov, E., Pfeiffer, R., Barajas-Martinez, H., Delpon, E., Hu, D., Desai, M. et al. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm 7, 1872–1882 (2010).
Fukuyama, M., Ohno, S., Wang, Q., Shirayama, T., Itoh, H. & Horie, M. Nonsense-mediated mRNA decay due to a CACNA1C splicing mutation in a patient with Brugada syndrome. Heart Rhythm 11, 629–634 (2014).
Delpon, E., Cordeiro, J. M., Nunez, L., Thomsen, P. E., Guerchicoff, A., Pollevick, G. D. et al. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ. Arrhythm. Electrophysiol. 1, 209–218 (2008).
Verkerk, A. O., Wilders, R., Schulze-Bahr, E., Beekman, L., Bhuiyan, Z. A., Bertrand, J. et al. Role of sequence variations in the human ether-a-go-go-related gene (HERG, KCNH2) in the Brugada syndrome. Cardiovasc. Res. 68, 441–453 (2005).
Wang, Q., Ohno, S., Ding, W. G., Fukuyama, M., Miyamoto, A., Itoh, H. et al. Gain-of-function KCNH2 mutations in patients with Brugada syndrome. J. Cardiovasc. Electrophysiol. 25, 522–530 (2014).
Medeiros-Domingo, A., Tan, B. H., Crotti, L., Tester, D. J., Eckhardt, L., Cuoretti, A. et al. Gain-of-function mutation, S422L, in the KCNJ8-encoded cardiac K ATP channel Kir6.1 as a pathogenic substrate for J wave syndromes. Heart Rhythm 7, 1466–1471 (2010).
Ueda, K., Hirano, Y., Higashiuesato, Y., Aizawa, Y., Hayashi, T., Inagaki, N. et al. Role of HCN4 channel in preventing ventricular arrhythmia. J. Hum. Genet. 54, 115–121 (2009).
Giudicessi, J. R., Ye, D., Tester, D. J., Crotti, L., Mugione, A., Nesterenko, V. V. et al. Transient outward current (I(to)) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm 8, 1024–1032 (2011).
Ohno, S., Zankov, D. P., Ding, W. G., Itoh, H., Makiyama, T., Doi, T. et al. KCNE5 (KCNE1L) variants are novel modulators of Brugada syndrome and idiopathic ventricular fibrillation. Circ. Arrhythm. Electrophysiol. 4, 352–361 (2011).
Boczek, N. J., Ye, D., Johnson, E. K., Wang, W., Crotti, L., Tester, D. J. et al. Characterization of SEMA3A-encoded semaphorin as a naturally occurring Kv4.3 protein inhibitor and its contribution to Brugada syndrome. Circ. Res. 115, 460–469 (2014).
Hu, D., Barajas-Martinez, H., Terzic, A., Park, S., Pfeiffer, R., Burashnikov, E. et al. ABCC9 is a novel Brugada and early repolarization syndrome susceptibility gene. Int. J. Cardiol. 171, 431–442 (2014).
Barajas-Martinez, H., Hu, D., Ferrer, T., Onetti, C. G., Wu, Y., Burashnikov, E. et al. Molecular genetic and functional association of Brugada and early repolarization syndromes with S422L missense mutation in KCNJ8. Heart Rhythm 9, 548–555 (2012).
Poelzing, S., Forleo, C., Samodell, M., Dudash, L., Sorrentino, S., Anaclerio, M. et al. SCN5A polymorphism restores trafficking of a Brugada syndrome mutation on a separate gene. Circulation (2006) 114, 368–376.
Bezzina, C. R., Barc, J., Mizusawa, Y., Remme, C. A., Gourraud, J. B., Simonet, F. et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet. 45, 1044–1049 (2013).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Watanabe, H., Minamino, T. Genetics of Brugada syndrome. J Hum Genet 61, 57–60 (2016). https://doi.org/10.1038/jhg.2015.97
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.97
This article is cited by
-
Novel SCN5A variants identified in a group of Iranian Brugada syndrome patients
Functional & Integrative Genomics (2021)
-
Brugada-Syndrom
Herzschrittmachertherapie + Elektrophysiologie (2020)
-
2019 HRS/EHRA/APHRS/LAHRS expert consensus statement on catheter ablation of ventricular arrhythmias
Journal of Interventional Cardiac Electrophysiology (2020)
-
Association between SCN5A and sudden unexplained nocturnal death syndrome in Thai decedents: a case–control study
Egyptian Journal of Forensic Sciences (2019)
