
Abstract
The arms of star-like nanogels were replaced by means of dynamic covalent exchange processes, and arm detachment and arm exchange reactions were successfully accomplished. Each star-like nanogel with alkoxyamine units at its branching points consisted of arms of poly(methyl methacrylate) (PMMA) synthesized by atom transfer radical polymerization, and a core of poly(divinylbenzene), synthesized by nitroxide-mediated radical polymerization using functionalized PMMA as a macroinitiator. The alkoxyamine groups were capable of serving as radically exchangeable units. Arm detachment reactions were carried out through a radical crossover reaction by heating the star-like nanogels with an excess of a small molecule alkoxyamine, whereas arm exchange reactions were performed by heating nanogels with higher molecular weight PMMA functionalized with alkoxyamine units at the chain ends. The molecular weights and sizes of the star-like nanogels were examined by gel permeation chromatography–multiangle light scattering and small-angle X-ray scattering measurements, respectively, and the shapes of nanogels were characterized by means of scanning force microscopy.
Similar content being viewed by others


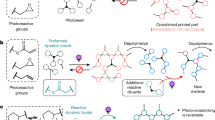
Introduction
Polymer gels have attracted considerable attention because their crosslinked structures permit the uptake of small molecules such as solvents, a process that has many applications in materials science and engineering. From the point of view of their crosslinked structures, gels can be divided into two main groups: chemical gels and physical gels. The crosslinking points of chemical gels consist of covalent bonds, whereas those of physical gels consist of non-covalent bonds. One advantage of physical gels is that their structure can be changed relatively easily; moreover, functional gels, such as self-healing cross-linked polymers,1, 2 have been developed. Recently, ‘reorganizable chemical gels’ in which the crosslinking structures change in response to specific external stimuli have been developed.3 Several functionalities, such as sol–gel transitions3, 4, 5 and controlled insertion of monomer units into crosslinking points,3 have been reported. Note that the concept of ‘dynamic covalent (bond) chemistry’,6, 7 which deals with equilibrium states of compounds based on covalently bonded systems, has contributed to the development of reorganizable chemical gels.
Microscopic gels have also been widely studied,8 and nanogels, in particular, are interesting research objects from the point of view of their applications.9, 10, 11 According to IUPAC,12 a nanogel is defined as a particle of gel of any shape with an equivalent diameter of approximately 1–100 nm. Several types of nanogels have been synthesized, such as poly(N-isopropylacrylamide) nanogels13 and cholesterol-bearing pullulan nanogels.14, 15 Crosslinked micelle or core crosslinked star polymers (star-like nanogels), which consist of a crosslinked core (nanogel) with several attached chains, have also been studied; these have been prepared by arm-first methods,16, 17, 18, 19 core-first methods,20, 21 micelle crosslinking methods22, 23 and others. The concept of reorganizable chemical gels has been applied to such star-like nanogels as well, and de-crosslinking reactions have been successfully accomplished by the use of disulfide bonds24 or ester bonds.25, 26 Furthermore, arm detachment reactions through ozonolysis22 or the use of cleavable bonds, such as ester bonds26, 27 or disulfide bonds,28 has also been reported.
Reorganizable chemical nanogels have also been developed using alkoxyamine compounds.29 Here, the term ‘alkoxyamine compounds’ is used as a generic term for compounds containing C–ON bonds. Although alkoxyamine compounds consisting of a styryl radical and a 2,2,6,6-tetramethylpiperidinyl-1-oxyl derivative behave as typical covalently bonded compounds, they dissociate on heating into their component radicals, and two characteristic reactions can occur. One is polymerization of styrene or acrylate derivatives, a process known as nitroxide-mediated radical polymerization (NMRP),30 and the other is the exchange reaction of the radical units, a process known as ‘dynamic covalent exchange’.31, 32 We have previously applied this chemistry to star-like nanogels and have reported the formation of star-like nanogels33, 34, 35 and structural transformations of these star-like nanogels into diblock copolymers by means of dynamic covalent exchange reactions.36, 37
Here, we propose a novel concept of ‘reorganizable star-like nanogels’, in which arm detachment and arm exchange reactions of star-like nanogels are performed by means of dynamic covalent exchange processes, as shown in Figure 1. Star-like nanogels with alkoxyamine units at their branching points were prepared by a combination of atom transfer radical polymerization (ATRP) and NMRP techniques, and arm detachment and arm exchange reactions were carried out by means of dynamic exchange reactions with alkoxyamine compounds and linear polymer chains with alkoxyamine end groups, respectively.

Schematic representation of an arm-replaceable star-like nanogel with alkoxyamine units, and its arm detachment and arm exchange reactions through dynamic covalent exchange processes. PDVB, poly(divinylbenzene); PMMA, poly(methyl methacrylate).
Experimental procedure
Materials
2,2,6,6-Tetramethyl-1-(1-phenylethoxy) piperidin-4-ol31 and 4-methoxy-1-((1′-phenylethyl)oxy)-2,2,6,6-tetramethylpiperidine (4)38 were prepared and purified as previously reported. Cu(I)Br (99+%) was purchased from Wako Pure Chemical Industries (Tokyo, Japan) and purified by stirring in acetic acid (Wako Pure Chemical Industries, 99+%), washing with ethanol (Wako Pure Chemical Industries, 99+%) and then drying in vacuo. 2-Bromoisobutyryl bromide (BrCMe2COBr, 98%), 4,4′-dinonyl-2,2′-bipyridine (97%) and tributyltin hydride (Bu3SnH, 97%) were purchased from Sigma-Aldirich (St Louis, MO, USA), and used without further purification. Triethyl amine ((Et)3N, 99%), methyl methacrylate (MMA, 99%), anisole (99%) and divinyl benzene (DVB) were purchased from Wako Pure Chemical Industries, and purified by distillation under reduced pressure over calcium hydrides. Tetrahydrofuran (THF, 99.5%) and toluene (99.5%) were purchased from Wako Pure Chemical Industries and purified with Glass Contour solvent purification systems (Glass Contour Solvent Systems, Nashua, NH, USA).
Measurements
1H (300 MHz) and 13C (75 MHz) nuclear magnetic resonance (NMR) spectroscopic measurements were taken at 25 °C with a JEOL JNM-AL300 spectrometer (JEOL, Tokyo, Japan) using tetramethylsilane as internal standard in chloroform-d (CDCl3). Monomer conversion was determined by 1H-NMR spectroscopy of the crude reaction mixtures. Infrared spectra were obtained with a Perkin-Elmer Spectrum One infrared spectrometer as thin films on NaCl.
Gel permeation chromatography (GPC) measurements were taken at 40 °C on a TOSOH HLC-8220 GPC system equipped with a guard column (TOSOH TSK guard column Super H-L, TOSOH, Yamaguchi, Japan), three columns (TOSOH TSK gel SuperH 6000, 4000 and 2500) and a ultraviolet-visible detector. THF was used as the eluent at a flow rate of 0.6 ml min−1. PSt standards (Mn=1060–1 090 000; Mw/Mn=1.02–1.08) were used for calibration to estimate number-average molecular weight (Mn) and molecular weight distribution (Mw/Mn) of the polymers.
Gel permeation chromatography–multiangle light scattering (GPC–MALS) measurements were taken in THF at 25 °C (column temperature: 40 °C) using a Dawn EOS instrument (Wyatt Technology, Santa Barbara, CA, USA; Ga-As laser, λ=690 nm) to evaluate absolute weight-average molecular weight (Mw, MALS) of the polymers. The specific refractive index increment (∂n/∂c), which is necessary for the analysis of GPC–MALS, was measured with Otsuka Electric DRM-3000 (Otsuka Electric, Osaka, Japan) (λ=633 nm) at 25 °C. The measured ∂n/∂c of star-like nanogel 3 (polymerized for 8 h) in THF was 0.119 cm3 g−1.
Small-angle X-ray scattering (SAXS) measurements were taken at the BL40B2 beam line in SPring-8 using an incident X-ray with λ=0.150 nm. Scattered X-rays were detected using a 300 × 300 mm imaging plate with a resolution of 0.1 mm/pixel and 2187 mm sample-to-detector distance calibrated by the average of 11 peaks of collagen. The measured samples were four concentrations of polymers in toluene contained in 2-mm-diameter quartz capillaries. The scattering intensities of polymers (ΔI(q)=Isoln(q)/Tsoln−Isolv(q)/Tsolv) were calculated by subtracting the scattering intensities of solvent (Isolv(q)) from that of the solution (Isoln(q)) adjusted through transmittance (Tsoln and Tsolv), and extrapolating to zero concentration of the four samples. The radius of gyration (Rg) of polymers was evaluated by initial slopes and intercepts of square-root plots ((c0/ΔI(q))1/2 vs q2).39
Scanning force microscopic (SFM) observations were made using a SPA400 (SII Nanotechnology, Chiba, Japan) in dynamic force mode. A 20 × 20 μm2 scanner and a cantilever (SI-DF20, resonance frequency ≈128 kHz, spring constant 13 Nm−1) were used with an amplitude set point of ≈0.9. The samples for the SFM observations were prepared by spin casting (1000 r.p.m., 10 s) of 1 × 10−4 wt% chloroform solutions of the polymers onto freshly cleaved mica.
2,2,6,6-tetramethyl-1-(1-phenylethoxy)piperidin-4-yl 2-bromo- 2-methylpropanoate (1)
A round-bottomed flask was charged with 2,2,6,6-tetramethyl-1-(1-phenylethoxy)piperidin-4-ol (0.832 g, 3.00 mmol) and dried under vacuum. The atmosphere in the flask was replaced with argon, and THF (7.5 ml) and Et3N (627 μl, 4.5 mmol) were added to the flask, followed by BrCMe2COBr (556 μl, 4.5 mmol) added dropwise from a syringe at 0 °C. The mixture was stirred for 12 h before MeOH (five drops) was added and the solvent was removed under vacuum. Water and dichloromethane were added and the separated aqueous layer was washed with dichloromethane. The combined organic layers were concentrated to yield a crude product that was purified by chromatography (silica gel, EtOAc–hexane (1:10 v/v)) and dried in vacuo to yield 1 as a colorless transparent oil; yield: 1.17 g (92%). 1H NMR: δ/p.p.m. 0.67 (s, 3H, CH3), 1.13 (s, 3H, CH3), 1.27 (s, 3H, CH3), 1.35 (s, 3H, CH3), 1.49 (d, J=7 Hz, 3H, CH3), 1.60 (d, J=12 Hz, 1H, CH2), 1.67 (d, J=12 Hz, 1H, CH2), 1.77 (dt, J=12 Hz, 4 Hz, 1H, CH2), 1.89 (s, 3H, CH3), 4.77 (q, J=7 Hz, 1H, CH), 5.03 (m, 1H, CH), 7.20–7.36 (m, 5H, aromatic). 13C NMR: δ/p.p.m. 19.01, 21.13, 28.56, 31.96, 32.27, 41.89, 41.96, 53.92, 57.83, 58.09, 66.57, 81.30, 124.61, 124.95, 125.99, 143.15, 169.15. Fourier transform infrared (Neat, cm−1): 2978 (C–H), 1727 (C=O), 1170, 908, 651. High-resolution mass spectrometer exact mass calculated for [M+1]+ C21H33NO3Br 426.1644, found 426.1683.
Alkoxyamine-terminated poly(methyl methacrylate) (2a)
A glass tube was charged with CuBr (14.4 mg, 0.1 mmol) and 4,4′-dinonyl-2,2′-bipyridine (81.63 mg, 0.2 mmol) and dried under vacuum. The atmosphere in the flask was replaced with argon, and argon-purged MMA (4.28 ml, 40 mmol) and anisole (4.3 ml) were added to the tube. The solution was then degassed by five freeze–pump–thaw cycles. Ester 1 (42.64 mg, 0.1 mmol) was added under argon, and the mixture was stirred for 15 h. The glass tube was immersed in ice water, and acetone was added. The solution was purified by filtration through an alumina column to remove the Cu catalyst, and the product was precipitated with MeOH. The precipitate was dried under vacuum to produce a white powder; yield: 2.51 g (63%); conversion: 72%; Mn=27 700; Mw/Mn=1.21. The C–Br bonds at the chain ends were hydrogenated, as previously described,40 using a 2,2′-azobisisobutyronitrile/Bu3SnH system in toluene at 45 °C.
A higher molecular weight analog of 2a (2b) was similarly prepared by bulk polymerization of MMA.
Core crosslinked polymer 3 (star-like nanogel) by the NMRP method
In a typical run, a glass tube was charged with the end-functionalized polymer 2a (138.5 mg, 0.005 mmol), DVB (71.2 μl, 0.5 mmol) and anisole (1.25 ml), and the solution was degassed by seven freeze–pump–thaw cycles. Subsequently, the glass tube was sealed under vacuum and heated at 125 °C for 8 h. The solvent was removed under vacuum, and the polymer was purified by freeze-drying with benzene to yield a white powder (conversion=56%). For GPC–MALS, SAXS and SFM studies, the resulting polymers were purified by preparative high-performance liquid chromatography (HPLC) to remove non-crosslinked poly(methyl methacrylate) (PMMA).
Arm detachment reaction
In a typical run, a glass tube was charged with core crosslinked polymer 3 (10 mg, 0.292 mmol for alkoxyamine units), 4-methoxy-2,2,6,6-tetramethyl-1-(1-phenylethoxy)piperidine (4; 34.1 mg, 117 mmol) and anisole (1 ml). The solution was degassed by seven freeze–pump–thaw cycles, and then the tube was sealed under vacuum and heated at 100 °C for 24 h. For GPC, SAXS and SFM studies, the resulting polymers were purified by preparative HPLC to remove unreacted 4.
Arm exchange reaction
In a typical run, a glass tube was charged with crosslinked polymer 3 (1 mg, 0.0292 mmol for alkoxyamine units), polymer 2b (49.8 mg, 0.585 mmol) and anisole (1.67 ml). The solution was degassed by seven freeze–pump–thaw cycles, and then the glass tube was sealed under vacuum and heated at 100 °C for 24 h. For GPC, SAXS and SFM studies, the resulting polymers were purified by preparative HPLC to remove unreacted 2b.
Results and Discussion
Molecular design and preparation of star-like nanogels
Star-like nanogels with alkoxyamine units at their branching points were prepared in an arm-first manner by sequential ATRP and NMRP using a bifunctional initiator. A shell-crosslinked micelle with alkoxyamine units at the core-shell interfaces has been previously reported and was prepared by ATRP and NMRP methods.29 First, MMA was polymerized by the ATRP method from an alkyl bromide group of the bifunctional initiator to form a linear polymer, and subsequently DVB, as the crosslinkable monomer, was polymerized from the alkoxyamine groups of the resulting macroinitiator by NMRP to form the core crosslinked star polymer (star-like nanogel). For the preparation of PMMA, the position of connection to the alkoxyamine unit (at a styryl unit site or a 2,2,6,6-tetramethylpiperidinyl-1-oxyl unit site) is critical, because the positions of the alkoxyamine units in the star-like nanogels are determined at this stage. When the PMMA chain is connected to the 2,2,6,6-tetramethylpiperidinyl-1-oxyl site, the corresponding star-like nanogels have alkoxyamine units at the connection points between the PMMA arms and the poly(divinylbenzene) core.41, 42 Had the PMMA chain been connected to a styryl radical site in the alkoxyamine unit, the corresponding star-like nanogels would have had alkoxyamine units at the chain ends of the cores,43, 44 and the arms could not then be replaced by dynamic covalent exchange processes. Taking this difference in molecular design into consideration, we synthesized bifunctional initiator 1 for ATRP and NMRP by condensation of 2-bromoisobutyryl bromide with 2,2,6,6-tetramethyl-1-(1-phenylethoxy)piperidin-4-ol, as shown in Scheme 1. The reaction proceeded quantitatively to afford ester 1, the structure of which was confirmed by NMR and infrared spectroscopy and by mass spectroscopic measurements.
The PMMA prepolymer with alkoxyamine units at the chain ends was synthesized by ATRP of MMA with a copper(I) bromide/4,4′-dinonyl-2,2′-bipyridine catalyst system, as shown in Scheme 1. The reaction was carried out at 50 °C, because the alkoxyamine units are stable under these conditions.32 Controlled polymerization was achieved, as both the plot of ln[M]0/[M] (where [M]0 is the initial concentration of monomer and [M] is the concentration of monomer in each reaction time) versus the reaction time and the plot of the number-average molecular mass (Mn) versus the conversion were linear. Two types of PMMA (2a and 2b), which had different molecular weights, were synthesized by changing the concentrations of the initiator and catalyst and performing the polymerization in solution or in the bulk phase, as listed in Table 1. The Mn of 2b was about three times that of 2a, and polymers with relatively narrow molecular weight distributions were obtained in both cases. The attachment of alkoxyamine units in 2 was confirmed by 1H-NMR measurements. After polymerization, the C–Br bonds in the chain ends of the PMMA prepolymers were subjected to hydrogenolysis40 to prevent side reactions from occurring in subsequent NMRP and dynamic covalent exchange reactions.
Star-like nanogels with alkoxyamine units at their branching points (3) were prepared by NMRP of crosslinkable DVB monomer from the linear PMMA macroinitiator 2a, which is similar to the usual method for forming core crosslinked star polymers.16 The polymerization was performed by heating 2a with DVB ([DVB]/[2a]=100:1) in anisole at 125 °C, as shown in Scheme 1. Most importantly, no macroscopic gelation was observed during the polymerization reaction. Figure 2a shows GPC curves for the reaction mixture at several reaction times. As the reaction time increased, new peaks in the higher molecular weight region appeared, and the intensity of these peaks increased, whereas those of the PMMA macroinitiator decreased. The course of the polymerization was followed by means of its kinetic behavior, and values of ln([M]0/[M]) and the weight-average molecular mass determined by MALS (Mw,MALS) at several reaction times are shown in Figure 2b. At times of up to 8 h, the plots of ln([M]0/[M]) increased linearly with time, showing that the concentration of radicals was constant during the polymerization reaction, thereby confirming that the polymerization proceeded by typical NMRP from the PMMA macroinitiator 2a. Indeed, the values of Mw,MALS increased markedly, confirming that crosslinked polymers were formed. Although the Mn,MALS estimated by MALS measurements is inexact values, the arm number (f=n × Mn,MALS(nanogel)/Mn(arm)) in 8 h was roughly calculated as f=18, where n is the weight fraction of the arm segment. The crosslinked polymer obtained by heating for 8 h was used for subsequent arm detachment and arm exchange reactions of the polymer.

(a) Gel permeation chromatography curves for the reaction mixtures after polymerization of divinyl benzene by nitroxide-mediated radical polymerization with the poly(methyl methacrylate) initiator 2a (Mn=27 100, Mw/Mn=1.21), and (b) temporal variations of Mw,MALS and ln[M]0/[M]. MALS, multiangle light scattering.
The sizes of the crosslinked polymers were investigated by means of SAXS measurements, and the values of the radius of gyration (Rg) were determined from the initial slopes and intercepts of the square-root plots of ((c/ΔI(q))1/2 versus q2.39 Figure 3a shows the temporal variations in Rg values, which increased with increasing reaction time. After heating for 8 h, the radius of gyration was estimated to be 25 nm. These results confirmed that nanosized crosslinked polymers were successfully synthesized by the ATRP and NMRP methods.

(a) Radius of gyration (Rg) of reaction products after polymerization of divinyl benzene by nitroxide-mediated radical polymerization using poly(methyl methacrylate) macroinitiator 2a at several reaction times, and (b) a dynamic mode scanning force micrograph of the polymer prepared by polymerization for 8 h and cast on a mica substrate.
SFM, which is one of the most useful tools for direct observation of polymer structures,45, 46 was used to examine whether star-like nanogel architectures were formed. SFM observations were performed by using dynamic force modes on samples prepared by spin casting a chloroform solution of nanogels onto a mica substrate. Figure 3b shows an SFM image of star-like nanogel 3 prepared with an 8 h polymerization time. The image confirms the presence of a core with several connecting chains. The size of the core, estimated from the average of 50 images, was 5.0 nm in height and 29.8 nm in diameter, showing that the core was flattened on the mica substrate.
Arm detachment reaction
The arm detachment reaction of the star-like nanogels was performed by a means of a dynamic covalent exchange reaction47 between the alkoxyamine units at the branching points of star-like nanogels and additional excess alkoxyamine 4. Before the arm detachment reaction, the star-like nanogels were purified by preparative HPLC to remove non-crosslinked PMMA components. Star-like nanogel 3, obtained after an 8 h reaction, was heated at 100 °C in anisole solution with an excess of alkoxyamine 4 (an equivalent of 400, based on the alkoxyamine present at branching points), as shown in Figure 1. Chloroform-insoluble components began to precipitate after several hours, and their amounts increased as the reaction proceeded up to 8 h.
Figure 4a shows the GPC curves for the reaction mixtures at several reaction times. The molecular weights corresponding to the peaks decreased, and new peaks appeared and gradually grew; the molecular weight corresponding to the new peaks was almost the same as that of PMMA 2a (refer to 0 h in Figure 2a). The peaks with higher molecular weights, shown as component (i) in Figure 4a, correspond to a mixture of completely and incompletely detached nanogels, which means that not all arms were detached from nanogels even after 24 h. The peaks (i) were fractionated by preparative HPLC using chloroform as an eluent. Because poly(divinylbenzene) was insoluble in chloroform, some insoluble matter precipitated. The remaining soluble components were subjected to SAXS measurements in toluene, and their Rg values were evaluated as shown in Figure 4b. As the reaction proceeded, the Rg values decreased, indicating that the arms were detached and that their sizes decreased. Indeed, the low molecular weight component (ii) was also examined by SAXS. The Rg of this fraction after 24 h was 6.4 nm (in the case of 2a, Rg=4.3 nm), and the scattering profile in the SAXS measurement corresponded closely to the scattering function for a Gaussian chain. In addition, the peak was confirmed as PMMA by an 1H-NMR measurement. These results clearly show that arm detachment reaction proceeded, with formation of poly(divinylbenzene) and linear PMMA.

(a) Gel permeation chromatography curves of reaction mixtures after heating nanogels 3 with excess alkoxyamine 4 (equivalent to 400, based on the alkoxyamine units in the nanogels), and (b) radius of gyration (Rg) of the high molecular weight component (i) after various reaction times.
Figure 5 shows an SFM image of nanogels 3 after heating with excess of 4 for 24 h, without HPLC fractionation. Two round-shaped structures and some linear-shaped structures were observed; these must correspond to the poly(divinylbenzene) nanogels after detachment of the PMMA arms and the detached linear PMMA chains, respectively. This SFM image also confirms that the arm detachment reaction proceeded successfully. However, the upper round-shaped structure seems to have retained a few connected chains, which may result in partial solubility of the nanogels. The core sizes before heating (5.0 nm in height and 29.8 nm in diameter) and after heating (4.9 nm in height and 30.6 nm in diameter), taken as average values for 50 images, were almost the same, showing that side reactions, such as a coupling of the cores, barely occurred during this reaction.

A scanning force microscopic height image of the reaction mixture after heating nanogels 3 with excess alkoxyamine 4 (equivalent to 400, based on the alkoxyamine units in the nanogels) for 24 h.
Arm exchange reaction
The arm exchange reaction was performed by a dynamic covalent exchange between the alkoxyamine units in the star-like nanogels 3 and alkoxyamine-terminated higher molecular weight PMMA 2b. The reaction was carried out by heating nanogels 3 with excess polymer 2b (equivalent of 20, based on the alkoxyamine units in the nanogel) at 100 °C in anisole, as shown in Figure 1; the molecular weight of 2b was three times that of 2a. To permit detection of any changes in the molecular weight of star-like nanogels in the presence of excess PMMA, a ultraviolet-visible detector was used for the GPC measurements. Figure 6a shows the GPC curves for the reaction mixtures after several reaction times. The molecular weights of the peaks increased with increasing reaction time. Furthermore, the Rg values in toluene, estimated by SAXS measurements, increased as the reaction proceeded, as shown in Figure 6b. These results confirmed that arm exchange reactions involving substitution of PMMA arms 2a with arms of PMMA 2b had occurred.

(a) Gel permeation chromatography curves of the reaction mixture after heating 3 with poly(methyl methacrylate) 2b (equivalent to 20, based on the alkoxyamine units in 3), as detected by a ultraviolet-visible detector, and (b) the corresponding radius of gyration (Rg) values at several reaction times.
In the arm exchange reaction, the structure of the star-like nanogels would be expected to change markedly, and this was investigated by microscopy. Figure 7a shows an SFM image of the product mixture of the arm exchange reaction after heating for 24 h. An image consisting of a core and several connecting chains with long arms and short arms was observed because the molecular weight of 2b was three times that of 2a. Figure 7b shows the core sizes (in height and diameter) and the number ratio of long arms to total arms of the product mixtures after several reaction times. The core size of the polymers did not change during the reaction, indicating that coupling reactions between cores barely occurred. In contrast, the number ratio of long arms to all arms increased with increasing reaction time. However, a residue of short chains remained, even after 24 h. This is probably the result of steric hindrance to the longer intricate PMMA chains. These results provide strong confirmation that arm exchange reactions proceeded and that hetero arm star-like nanogels with different arm lengths were formed.

(a) Dynamic mode scanning force microscopic image on mica of star-like nanogels after heating 3 with 2b for 24 h in anisole, and (b) core size in height and diameter, and number ratio of long arms to total arms of star-like nanogels after the arm exchange reaction.
Conclusions
We have demonstrated the concept of arm-replaceable star-like nanogels and have accomplished arm detachment and arm exchange reactions. The star-like nanogels were synthesized by a two-step polymerization process. PMMA terminated by alkoxyamine units was prepared by ATRP, and alkoxyamine units subsequently served as macroinitiators for polymerization of DVB by NMRP. In the polymerization of DVB, the reaction proceeded by NMRP from the viewpoint of its kinetic behavior. An arm detachment reaction was carried out by heating star-like nanogels with an excess of a low molecular weight alkoxyamine to induce dynamic covalent exchange. GPC curves showed the presence of peaks corresponding to the PMMA chains, and the presence of arm detached nanogels (cores) and linear polymer chains (arms) was confirmed by SFM observation. An arm exchange reaction was also performed by dynamic covalent exchange between the star-like nanogels and high molecular weight PMMA terminated with alkoxyamine units; the molecular weights and sizes increased with increasing reaction time. Furthermore, the formation of hetero arm star-like nanogels was confirmed by SFM observations, which showed that the arm exchange reaction had proceeded successfully. This novel concept of reorganizable chemical nanogels could be used to produce stimuli-responsive star-like nanogels, as well as to build systems with more complicated macromolecular designs.

Synthesis of bifunctional initiator 1, preparation of poly(methyl methacrylate) 2 with a terminal alkoxyamine unit by atom transfer radical polymerization method, and preparation of star-like nanogel 3 via nitroxide-mediated radical polymerization of divinyl benzene. THF, tetrahydrofuran.
References
Cordier, P., Tournilhac, F., Soulie-Ziakovic, C. & Leibler, L. Self-healing and thermoreversible rubber from supramolecular assembly. Nature 451, 977–980 (2008).
Wang, Q., Mynar, J. L., Yoshida, M., Lee, E., Lee, M., Okuro, K., Kinbara, K. & Aida, T. High-water-content mouldable hydrogels by mixing clay and a dendritic molecular binder. Nature 463, 339–343 (2010).
Amamoto, Y., Kikuchi, M., Masunaga, H., Sasaki, S., Otsuka, H. & Takahara, A. Reorganizable chemical polymer gels based on dynamic covalent exchange and controlled monomer insertion. Macromolecules 42, 8733–8738 (2009).
Deng, G. H., Tang, C. M., Li, F. Y., Jiang, H. F. & Chen, Y. M. Covalent cross-linked polymer gels with reversible sol-gel transition and self-healing properties. Macromolecules 43, 1191–1194 (2010).
Higaki, Y., Otsuka, H. & Takahara, A. A thermodynamic polymer cross-linking system based on radically exchangeable covalent bonds. Macromolecules 39, 2121–2125 (2006).
Rowan, S. J., Cantrill, S. J., Cousins, G. R. L., Sanders, J. K. M. & Stoddart, J. F. Dynamic covalent chemistry. Angew. Chem. Int. Ed. 41, 898–952 (2002).
Maeda, T., Otsuka, H. & Takahara, A. Dynamic covalent polymers: reorganizable polymers with dynamic covalent bonds. Prog. Polym. Sci. 34, 581–604 (2009).
Suzuki, D. & Yoshida, R. Self-oscillating core/shell microgels: effect of a crosslinked nanoshell on autonomous oscillation of the core. Polym. J. 42, 501–508 (2010).
Gota, C., Okabe, K., Funatsu, T., Harada, Y. & Uchiyama, S. Hydrophilic fluorescent nanogel thermometer for intracellular thermometry. J. Am. Chem. Soc. 131, 2766–2767 (2009).
Mattsson, J., Wyss, H. M., Fernandez-Nieves, A., Miyazaki, K., Hu, Z., Reichman, D. R. & Weitz, D. A. Soft colloids make strong glasses. Nature 462, 83–86 (2009).
Oh, J. K., Drumright, R., Siegwart, D. J. & Matyjaszewski, K. The development of microgels/nanogels for drug delivery applications. Prog. Polym. Sci. 33, 448–477 (2008).
Alemán, J., Chadwick, A. V., He, J., Hess, M., Horie, K., Jones, R. G., Kratochvíl, P., Meisel, I., Mita, I., Moad, G., Penczek, S., Stepto, R. F. T. & Jones, R. G. Definitions of terms relating to the structure and processing of sols, gels, networks, and inorganic-organic hybrid materials (IUPAC Recommendations 2007). Pure Appl. Chem. 79, 1801–1827 (2007).
Pelton, R. Temperature-sensitive aqueous microgels. Adv. Colloid Interfac. 85, 1–33 (2000).
Akiyoshi, K., Deguchi, S., Moriguchi, N., Yamaguchi, S. & Sunamoto, J. Self-aggregates of hydrophobized polysaccharides in water—formation and characteristics of nanoparticles. Macromolecules 26, 3062–3068 (1993).
Nishikawa, T., Akiyoshi, K. & Sunamoto, J. Macromolecular complexation between bovine serum albumin and the self-assembled hydrogel nanoparticle of hydrophobized polysaccharides. J. Am. Chem. Soc. 118, 6110–6115 (1996).
Blencowe, A., Tan, J. F., Goh, T. K. & Qiao, G. G. Core cross-linked star polymers via controlled radical polymerisation. Polymer 50, 5–32 (2009).
Bosman, A. W., Vestberg, R., Heumann, A., Frechet, J. M. J. & Hawker, C. J. A modular approach toward functionalized three-dimensional macromolecules: from synthetic concepts to practical applications. J. Am. Chem. Soc. 125, 715–728 (2003).
Gao, H. F. & Matyjaszewski, K. Synthesis of functional polymers with controlled architecture by CRP of monomers in the presence of cross-linkers: from stars to gels. Prog. Polym. Sci. 34, 317–350 (2009).
Terashima, T., Ouchi, M., Ando, T., Kamigaito, M. & Sawamoto, M. Amphiphilic, thermosensitive ruthenium(II)-bearing star polymer catalysts: one-pot synthesis of PEG armed star polymers with ruthenium(II)-enclosed microgel cores via metal-catalyzed living radical polymerization. Macromolecules 40, 3581–3588 (2007).
Gao, H. F. & Matyjaszewski, K. Synthesis of star polymers by a new ‘Core-First’ method: sequential polymerization of cross-linker and monomer. Macromolecules 41, 1118–1125 (2008).
Goh, T. K., Sulistio, A. P., Blencowe, A., Johnson, J. W. & Qiao, G. G. Synthesis and characterization of core cross-linked star clusters by conventional free-radical polymerization. Macromolecules 40, 7819–7826 (2007).
Fujii, S., Cai, Y. L., Weaver, J. V. M. & Armes, S. P. Syntheses of shell cross-linked micelles using acidic ABC triblock copolymers and their application as pH-responsive particulate emulsifiers. J. Am. Chem. Soc. 127, 7304–7305 (2005).
Sakellariou, G., Avgeropoulos, A., Hadjichristidis, N., Mays, J. W. & Baskaran, D. Functionalized organic nanoparticles from core-crosslinked poly(4-vinylbenzocyclobutene-b-butadiene) diblock copolymer micelles. Polymer 50, 6202–6211 (2009).
Oh, J. K., Tang, C. B., Gao, H. F., Tsarevsky, N. V. & Matyjaszewski, K. Inverse miniemulsion ATRP: a new method for synthesis and functionalization of well-defined water-soluble/cross-linked polymeric particles. J. Am. Chem. Soc. 128, 5578–5584 (2006).
Wiltshire, J. T. & Qiao, G. G. Degradable core cross-linked star polymers via ring-opening polymerization. Macromolecules 39, 4282–4285 (2006).
Wiltshire, J. T. & Qiao, G. G. Selectively degradable core cross-linked star polymers. Macromolecules 39, 9018–9027 (2006).
Liu, C., Zhang, Y. & Huang, J. L. Well-defined star polymers with mixed-arms by sequential polymerization of atom transfer radical polymerization and reverse addition-fragmentation chain transfer on a hyperbranched polyglycerol core. Macromolecules 41, 325–331 (2008).
Zhang, M., Ishii, A., Nishiyama, N., Matsumoto, S., Ishii, T., Yamasaki, Y. & Kataoka, K. PEGylated calcium phosphate nanocomposites as smart environment-sensitive carriers for siRNA delivery. Adv. Mater 21, 3520–3525 (2009).
Murthy, K. S., Ma, Q. G., Clark, C. G., Remsen, E. E. & Wooley, K. L. Fundamental design aspects of amphiphilic shell-crosslinked nanoparticles for controlled release applications. Chem. Commun. 773–774 (2001).
Georges, M. K., Veregin, R. P. N., Kazmaier, P. M. & Hamer, G. K. Narrow molecular-weight resins by a free-radical polymerization process. Macromolecules 26, 2987–2988 (1993).
Hawker, C. J., Barclay, G. G. & Dao, J. L. Radical crossover in nitroxide mediated ‘living’ free radical polymerizations. J. Am. Chem. Soc. 118, 11467–11471 (1996).
Otsuka, H., Aotani, K., Higaki, Y. & Takahara, A. A dynamic (reversible) covalent polymer: radical crossover behaviour of TEMPO-containing poly(alkoxyamine ester)s. Chem. Commun. 2838–2839 (2002).
Amamoto, Y., Higaki, Y., Matsuda, Y., Otsuka, H. & Takahara, A. Programmed formation of nanogels via a radical crossover reaction of complementarily reactive diblock copolymers. Chem. Lett. 36, 774–775 (2007).
Amamoto, Y., Kikuchi, M., Otsuka, H. & Takahara, A. Solvent-controlled formation of star-like nanogels via dynamic covalent exchange of PSt-b-PMMA diblock copolymers with alkoxyamine units in the side chain. Macromolecules 43, 5470–5473 (2010).
Amamoto, Y., Maeda, T., Kikuchi, M., Otsuka, H. & Takahara, A. Rational approach to star-like nanogels with different arm lengths: formation by dynamic covalent exchange and their imaging. Chem. Commun. 689–691 (2009).
Amamoto, Y., Higaki, Y., Matsuda, Y., Otsuka, H. & Takahara, A. Programmed thermodynamic formation and structure analysis of star-like nanogels with core cross-linked by thermally exchangeable dynamic covalent bonds. J. Am. Chem. Soc. 129, 13298–13304 (2007).
Amamoto, Y., Kikuchi, M., Masunaga, H., Sasaki, S., Otsuka, H. & Takahara, A. Intelligent build-up of complementarily reactive diblock copolymers via dynamic covalent exchange toward symmetrical and miktoarm star-like nanogels. Macromolecules 43, 1785–1791 (2010).
Higaki, Y., Otsuka, H. & Takahara, A. Dynamic formation of graft polymers via radical crossover reaction of alkoxyamines. Macromolecules 37, 1696–1701 (2004).
Nakamura, Y., Sugiyama, M., Amitani, K. & Norisuye, T. Synchrotron small-angle X-ray scattering from polystyrene polymacromonomers in toluene. Polym. J. 39, 1098–1104 (2007).
Coessens, V. & Matyjaszewski, K. Dehalogenation of polymers prepared by atom transfer radical polymerization. Macromol. Rapid. Comm. 20, 66–70 (1999).
Ruehl, J. & Braslau, R. A bidirectional ATRP-NMRP initiator: the effect of nitroxide size on the rate of nitroxide-mediated polymerization. J. Polym. Sci. Part A: Polym. Chem. 45, 2015–2025 (2007).
Shi, Y., Fu, Z. F., Li, B. Y. & Yang, W. T. Synthesis of diblock copolymers by combining stable free radical polymerization and atom transfer radical polymerization. J. Polym. Sci. Part A: Polym. Chem. 44, 2468–2475 (2006).
Tang, C. B., Kowalewski, T. & Matyjaszewski, K. Preparation of polyacrylonitrile-block-poly(n-butyl acrylate) copolymers using atom transfer radical polymerization and nitroxide mediated polymerization processes. Macromolecules 36, 1465–1473 (2003).
Tunca, U., Karliga, B., Ertekin, S., Ugur, A. L., Sirkecioglu, O. & Hizal, G. Synthesis of asymmetric difunctional initiators and their use in the preparation of block copolymers via ATRP and SFRP. Polymer 42, 8489–8493 (2001).
Kumaki, J., Nishikawa, Y. & Hashimoto, T. Visualization of single-chain conformations of a synthetic polymer with atomic force microscopy. J. Am. Chem. Soc. 118, 3321–3322 (1996).
Matyjaszewski, K., Qin, S. H., Boyce, J. R., Shirvanyants, D. & Sheiko, S. S. Effect of initiation conditions on the uniformity of three-arm star molecular brushes. Macromolecules 36, 1843–1849 (2003).
Jackson, A. W. & Fulton, D. A. Dynamic covalent diblock copolymers prepared from RAFT generated aldehyde and alkoxyamine end-functionalized polymers. Macromolecules 43, 1069–1075 (2010).
Acknowledgements
We gratefully acknowledge the financial support of a Grant-in-Aid for Scientific Research (20350057) from the Ministry of Education, Culture, Science, Sports and Technology of Japan. This work is also supported by a Grant-in-Aid for the Global COE Program, ‘Science for Future Molecular Systems’ from the Ministry of Education, Culture, Science, Sports and Technology of Japan. YA acknowledges the financial support of a Grant-in-Aid for JSPS Fellows. Drs Sono Sasaki and Hiroyasu Masunaga of Japan Synchrotron Radiation Research Institute are acknowledged for their help on SAXS measurements. The synchrotron radiation experiments were conducted at a BL40B2 in SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (proposal no. 2009A1011).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Amamoto, Y., Kikuchi, M., Otsuka, H. et al. Arm-replaceable star-like nanogels: arm detachment and arm exchange reactions by dynamic covalent exchanges of alkoxyamine units. Polym J 42, 860–867 (2010). https://doi.org/10.1038/pj.2010.83
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pj.2010.83
Keywords
This article is cited by
-
Visualization of judgment regions in convolutional neural networks for X-ray diffraction and scattering images of aliphatic polyesters
Polymer Journal (2021)
-
Reorganization of polymer structures based on dynamic covalent chemistry: polymer reactions by dynamic covalent exchanges of alkoxyamine units
Polymer Journal (2013)
