
Abstract
To introduce a phosphorylcholine (PC) moiety into segmented polyurethane, which has been widely used as an elastic material, the synthesis of a novel diol monomer containing a PC unit was performed. PC-containing polyurethanes were then prepared by polyaddition of the diol monomer and poly(carbonate diol) with 4,4′-diphenylmethane diisocyanate. The obtained polyurethanes were soluble in aprotic polar solvents, such as N,N-dimethylacetamide, N,N-dimethylformamide (DMF) and dimethyl sulfoxide, but were insoluble in water and alcohol. According to thermal analysis and the stress–strain measurements, the polyurethane films exhibited thermal stability up to 250 °C and elastic properties with high tensile strength. In addition, it was confirmed from the results of the protein adsorption experiments that the PC-containing polyurethane films efficiently reduced protein adsorption to the surface.
Similar content being viewed by others

Introduction
Polyurethane elastomers have been widely used in practical applications for medical devices1, 2 owing to their high mechanical strength and biocompatibility. Segmented polyurethanes (SPUs) generally consist of short alternating blocks of soft and hard segments. The soft segment is typically a component with a low glass transition temperature, such as aliphatic polyether, polyester and polycarbonate oligomers, the molecular weights of which are 400–5000 g mol−1. The hard segment is generally a component with a high glass transition temperature, such as aromatic diisocyanate linked to a low-molecular weight chain extender. The biocompatibility of SPU is thought to arise from the microphase separation of the soft and hard segments. However, the biostability of SPU is not suitable for long-term implantation. It has been suggested that the biodegradation and cracking of polyurethane that occurred in vivo was due to the adsorption of proteins, adhesion of macrophages and peroxide formation,3, 4, 5 which resulted in the reduction of the mechanical strength of SPU. Moreover, the soft segment of SPU was reportedly degraded by oxygen radicals produced by adherent macrophages.6 Therefore, several studies of surface- or chemically modified SPUs have been conducted to improve biostability by reducing the adhesion of cells and proteins.7, 8, 9, 10, 11 Recently, Khan et al.12, 13 reported that a potential application of poly(carbonate-urethane) was as a long-term biomedical implant material due to its resistance to biodegradation and its biocompatibility.
In addition, the 2-methacryloyloxyethyl phosphorylcholine (MPC) polymer has been developed by Ishihara et al.14 as an excellent biocompatible material, which efficiently reduces the adhesion of cells and proteins to the polymer surface.15 The design of the MPC polymer was inspired by the chemical structure of the phospholipid polar group in biomembranes. In recent years, the MPC polymer has been widely applied in biological and medical fields. Ishihara and colleagues have also investigated a polymer composite consisting of SPU and MPC polymer to reduce protein adsorption to the polymer surface and to improve the biocompatibility of SPU.16, 17, 18, 19, 20, 21
In our previous studies, synthesis of diamine monomer containing the phosphorylcholine (PC) group was performed to prepare polyamides and poly(urethane-urea)s containing PC groups,22, 23, 24, 25 the backbone component of which was mechanically strong and durable in solvents compared with the performance of MPC polymers. The synthesized polymers possessed the high molecular weight required to prepare tough films via a solvent casting method. On the basis of a platelet adhesion test, the synthesized polymers exhibited excellent biocompatibility, and adhesion of human platelets to the film surface was efficiently reduced.25 In addition, stress–strain measurements revealed that the poly(urethane-urea) films exhibited high elastic mechanical properties; the Young’s modulus increased with increasing PC content.24, 25
The aim of this study was to prepare another type of PC-containing polyurethane to create practical biomaterials, which exhibit the excellent biocompatibility, processability, thermal stability and mechanical strength of elastomers. Accordingly, we designed a new diol monomer-containing PC group, 2-[3,5-bis(2-hydroxyethoxy)benzoyloxy]ethyl phosphorylcholine; (BHPC). Cooper et al.26 reported that a PC-containing polyurethane could be prepared using glycerophosphorylcholine as a diol monomer, and would exhibit elastic and biocompatible properties. We have designed the BHPC molecule based on the concepts that BHPC would be more hydrophobic than glycerophosphorylcholine and easier to handle as a monomer for polycondensation or polyaddition, and that both of the primary hydroxyl groups of BHPC would make the polymer have a high molecular weight due to its higher reactivity than glycerophosphorylcholine with its secondary hydroxyl group. In this paper, the synthesis of BHPC and the preparations of high molecular weight SPUs by polyaddition of BHPC with other comonomers are described. Furthermore, the characteristics of the obtained polyurethanes are discussed; the solubility, thermal stability, surface and mechanical properties and the protein adsorption of the polymer films were investigated.
Experimental Procedure
Materials
COP (2-chloro-2-oxo-1,3,2-dioxaphospholane) was synthesized according to the literature27 and purified by distillation under reduced pressure, with a boiling point of 85–87 °C per 0.8 mm Hg (literature: 79 °C per 0.4 mm Hg). Trimethylamine was purchased from Sigma-Aldrich (Tokyo, Japan) and used as received. Tetrahydrofuran (THF) was distilled over sodium to remove the small amount of water. Triethylamine and acetonitrile were freshly distilled over calcium hydride. 1,4-Butanediol (BD) and 1,3-bis(2-hydroxyethyl)benzene were purchased from Tokyo Kasei Chemical (Tokyo, Japan), and used as received. 4,4′-Diphenylmethane diisocyanate (MDI) and poly(carbonate diol;)(PCD, Mn (number-average molecular weight)=1000, m=6) were kindly supplied by Nippon Polyurethane Industry (Tokyo, Japan) and Asahi Kasei Corporation (Tokyo, Japan), respectively. The MPC polymer, poly(MPC-co-butyl methacrylate), MPC content: 30 mol%, was kindly supplied by Professor Kazuhiko Ishihara from the University of Tokyo as a reference sample for protein adsorption. Other chemical reagents were used without further purification.
Synthesis of methyl 3,5-dihydroxybenzoate (1)
3,5-Dihydroxybenzoic acid (20.0 g, 130 mmol) was dissolved in 100 ml of methanol to which 3.3 ml of concentrated sulfuric acid was added. After refluxing for 18 h, the solution was neutralized by adding aqueous NaHCO3 solution. The mixture was then evaporated under vacuum and was extracted with ethyl acetate. The solvent was evaporated under reduced pressure to afford methyl 3,5-dihydroxybenzoate (1) as a white powder. Yield: 19.3 g (88.4%).
Proton nuclear magnetic resonance (1H-NMR), δ (400 MHz, dimethyl sulfoxide (DMSO)-d6, p.p.m.): 3.80 (3H, s), 6.44 (1H, t, J=2.4 Hz), 6.83 (2H, d, J=2.4 Hz), 9.53 (2H, s).
Infrared (IR), ν (KBr, cm−1): 3244 (−OH), 3088, 2997, 2951, 1690 (C=O), 1603, 1489, 1443, 1308, 1263, 1163, 1101, 997, 870, 766, 670.
Synthesis of 2-benzyloxyethyl tosylate (2)
Under an argon atmosphere, a solution of tosyl chloride (40.6 g, 213 mmol) in 210 ml of THF was slowly added to a solution of 2-benzyloxyethanol (25.2 ml, 178 mmol) in 180 ml of THF at 0 °C. The reaction mixture was stirred at room temperature overnight, and it was poured into an excess of distilled water. The mixture was extracted with chloroform, and the organic layer was dried over sodium sulfate. After the solvent had evaporated under reduced pressure, the product was washed by hexane to afford 2-benzyloxyethyl tosylate (2) as a white solid. Yield: 46.2 g (84.8%).
1H-NMR, δ (400 MHz, DMSO-d6, p.p.m.): 2.42 (3H, s), 3.60 (2H, t, J=2.2 Hz), 4.17 (2H, t, J=4.4 Hz), 4.42 (2H, s), 7.23–7.36 (5H, m), 7.42 (2H, d, J=8.3 Hz), 7.78 (2H, d, J=8.3 Hz).
IR, ν (KBr, cm−1): 3028, 2957, 2905, 2841, 2805, 1595, 1493, 1445, 1358, 1188, 1140, 1115, 1094, 1023, 1009, 924, 880, 806, 780, 735, 664.
Synthesis of methyl 3,5-bis(2-benzyloxyethoxy)benzoate (3)
Under an argon atmosphere, 1 (2.20 g, 13.2 mmol), 2 (8.09 g, 26.4 mmol) and K2CO3 (3.65 g, 26.4 mmol) were mixed in 48 ml of N,N-dimethylacetamide. After stirring overnight at 85 °C, the mixture was poured into an excess of distilled water. Then, the mixture was extracted with ethyl acetate, and the organic layer was dried over sodium sulfate. After the solvent had evaporated under reduced pressure, the product was purified by column chromatography on silica gel with hexane/ethyl acetate (2/1 by volume) to give methyl 3,5-bis(2-benzyloxyethoxy)benzoate (3) as a transparent liquid. Yield: 4.83 g (83.8%).
1H-NMR, δ (400 MHz, DMSO-d6, p.p.m.): 3.76 (4H, t, J=4.4 Hz), 3.84 (3H, s), 4.17 (6H, t, J=4.4 Hz), 4.56 (4H, s), 6.78 (1H, s), 7.10 (2H, s), 7.23–7.34 (10H, m).
IR, ν (KBr, cm−1): 3063, 3030, 2868, 1719 (C=O), 1595, 1443, 1323, 1237, 1171, 1105, 1069, 1026, 864, 766, 737, 698.
Synthesis of 3,5-bis(2-benzyloxyethoxy)benzoic acid (4)
A solution of NaOH (1.44 g, 36.0 mmol) in 44 ml of methanol was added to a solution of 3 (7.87 g, 18.0 mmol) in 18 ml of THF. After 3 h of reflux, the reaction mixture was poured into an excess of aqueous HCl solution. The reaction mixture was then stirred overnight at room temperature, and the solvents were evaporated to afford 3,5-bis(2-benzyloxyethoxy)benzoic acid (4) as a white solid. Yield: 7.03 g (92.5%).
1H-NMR, δ (400 MHz, DMSO-d6, p.p.m.): 3.76 (4H, t, J=4.4 Hz), 4.18 (4H, t, J=4.4 Hz), 4.72 (4H, s), 6.82 (1H, s), 7.07 (2H, s), 7.25-7.38 (10H, m), 13.0 (1H, bs).
IR, ν (KBr, cm−1): 3440 (−OH), 3030, 2940, 2868, 1699 (C=O), 1595, 1445, 1420, 1358, 1339, 1300, 1271, 1181, 1123, 1105, 1076, 1044, 897, 739, 698.
Synthesis of 2-(3,5-bis(2-benzyloxyethoxy)benzoyloxy)ethanol (5)
The compound 4 (3.82 g, 9.04 mmol), 2-bromoethanol (1.70 g, 13.7 mmol) and K2CO3 (2.50 g, 18.1 mmol) were mixed in 40 ml of N,N-dimethylacetamide. After stirring overnight at 85 °C, the solvent was evaporated and the mixture was poured into an excess of distilled water. The mixture was then extracted with ethyl acetate, and the organic layer was dried over sodium sulfate. After the solvent was evaporated, the product was purified by column chromatography on silica gel with hexane/ethyl acetate (1/1 by volume) to afford 2-(3,5-bis(2-benzyloxyethoxy)benzoyloxy)ethanol (5) as a transparent liquid. Yield: 2.98 g (70.1%).
1H-NMR, δ (400 MHz, DMSO-d6, p.p.m.) (Supplementary Information): 3.70 (2H, q, J=4.4 Hz), 3.77 (4H, t, J=4.4 Hz), 4.19 (4H, t, J=4.4 Hz), 4.25 (2H, t, J=4.9 Hz), 4.56 (4H, s), 4.97 (1H, t, J=5.9 Hz), 6.83 (1H, t, J=2.4 Hz), 7.12 (2H, d, J=2.4 Hz), 7.26-7.37 (10H, m).
IR, ν (KBr, cm−1): 3420 (−OH), 3030, 2868, 2359, 2342, 1717 (C=O), 1595, 1449, 1300, 1235, 1173, 1028, 862, 741, 698, 669.
Synthesis of 2-(3,5-bis(2-benzyloxyethoxy)benzoyloxy)ethyl phosphorylcholine (6)
Under an argon atmosphere, COP (1.67 ml, 18.6 mmol) was gradually added to a solution of 5 (4.32 g, 9.26 mmol) and triethylamine (2.6 ml, 18.6 mmol) dissolved in 56 ml of THF at 0 °C. After stirring at room temperature for 2 h, a white salt was precipitated in the reaction mixture. Then, the salt was filtered off and the solution obtained was evaporated under reduced pressure. Next, 48 ml of acetonitrile was added to the mixture, and the precipitated salt was filtered off again. After the solvent was evaporated, the residue was poured into distilled water and extracted with chloroform. The solvents of the organic layer were evaporated under reduced pressure to afford 2-[2-(3,5-bis(2-benzyloxyethoxy)benzoyloxy)ethyl]-2-oxo-1,3,2-dioxaphospholane as a pale yellow liquid.
The obtained 2-[2-(3,5-bis(2-benzyloxyethoxy)benzoyloxy)ethyl]-2-oxo-1,3,2-dioxaphospholane was dissolved in 92 ml of acetonitrile at 0 °C under an argon atmosphere, and trimethylamine (4.32 ml, 46.4 mmol) was added to the solution, after which the reaction vessel was sealed with a glass cap. After stirring at 60 °C for 13 h, the reaction mixture was evaporated under reduced pressure to afford 2-(3,5-bis(2-benzyloxyethoxy)benzoyloxy)ethyl phosphorylcholine (6) as a pale yellow liquid. Yield: 4.52 g (77.3% from compound 5).
1H-NMR, δ (400 MHz, DMSO-d6, p.p.m.) (Supplementary Information): 3.09 (9H, s), 3.47 (2H, m), 3.77 (4H, t, J=4.4 Hz), 3.94 (2H, m), 4.02 (2H, m), 4.20 (4H, t, J=4.4 Hz), 4.35 (2H, t, J=4.4 Hz), 4.56 (4H, s), 6.85 (1H, t, J=2.4 Hz), 7.11 (2H, d, J=2.4 Hz), 7.26-7.38 (10H, m).
IR, ν (KBr, cm−1): 3086, 3028, 2936, 2870, 1717 (C=O), 1597, 1447, 1346, 1323 (P=O), 1300, 1238, 1173, 1103, 1072, 968, 741.
Synthesis of 2-(3,5-bis(2-hydroloxyethoxy)benzoyloxy)ethyl phosphorylcholine (BHPC)
Next, 5% Pd on charcoal powder (0.61 g, 0.32 mmol by Pd) was suspended in a solution of 6 (4.51 g, 7.14 mmol), dissolved in 70 ml of methanol. The mixture was degassed under reduced pressure at −78 °C, and the vessel was filled with hydrogen gas at >760 mm Hg. After stirring for 72 h at 35 °C, the Pd on charcoal was filtered off by washing with methanol, and the solvent was evaporated under reduced pressure. The product was then dried in vacuo to afford 2-(3,5-bis(2-hydroloxyethoxy)benzoyloxy)ethyl phosphorylcholine (BHPC) as a white solid. Yield: 3.10 g (96.2%).
1H-NMR, δ (400 MHz, DMSO-d6, p.p.m.) (Supplementary Information): 3.12 (9H, s), 3.51 (2H, t, J=4.4 Hz), 3.70 (4H, t, J=3.4 Hz), 3.96 (2H, m), 3.99-4.05 (6H, m), 4.33 (2H, t, J=4.4 Hz), 5.10 (2H, t, J=5.37 Hz), 6.73 (1H, d, J=2.4 Hz), 7.13 (2H, d, J=2.4 Hz).
IR, ν (KBr, cm−1): 3379 (−OH), 3040, 2943, 2882, 1717 (C=O), 1597, 1447, 1373, 1346, 1323 (P=O), 1300, 1235, 1173, 1057, 968, 796, 768.
Preparations of SPUs containing PC units (SPUPC-10a, −20a, −30a, −50a)
BHPC (0.545 g, 1.21 mmol) was dissolved in 1.5 ml of DMSO under an argon atmosphere, and a solution containing MDI (3.05 g, 12.1 mmol) in 20 ml of DMSO was gradually added. After the mixture was stirred at 70 °C for 10 min, a solution of PCD (10.9 g, 10.9 mmol) in 11 ml of DMSO was added. Then, after the mixture was stirred at 70 °C for 1 h, the reaction mixture was poured into excess methanol to yield a white precipitate. The precipitate was collected by filtration and purified by reprecipitation from DMSO to excess methanol. Finally, the product was dried in vacuo to afford segmented polyurethane containing PC units (SPUPC-10a) as a pale yellow powder. Yield: 9.25 g (63.8%).
1H-NMR, δ (400 MHz, DMSO-d6, p.p.m.): 1.28 (-OCH2CH2CH2CH2CH2CH2OCO-, m), 1.55 (-OCH2CH2CH2CH2CH2CH2OCO-, m), 3.10 (-N+(CH3)3, m), 3.52 (-POCH2CH2-N, m), 3.75 (-Ph-CH2-Ph-, bs), 4.02 (-OCH2CH2OPh-, -OCH2CH2CH2CH2CH2CH2OCO-, m), 4.15 (-COOCH2CH2OP-, m), 4.24 (-POCH2CH2-N, m), 4.36 (-OCH2CH2OPh-, -COOCH2CH2OP-, m), 6.83 (-Ph-, m), 7.08 (-Ph-, m), 7.33 (-Ph-, m), 9.48 (-NHCOO-, bs), 9.70 (-NHCOO-, bs).
IR, ν (KBr, cm−1): 3340 (N-H), 2938, 2860, 1738 (C=O), 1597, 1530, 1464, 1404, 1381, 1310 (P=O), 1242, 1220, 1065, 1016, 914, 814, 770.
SPUPC-20a, SPUPC-30a and SPUPC-50a were prepared similarly to the procedure described above for SPUPC-10a (Supplementary Information), by changing the molar ratios of BHPC and PCD, as shown in Table 1.
Preparations of SPUs containing PC units (SPUPC-30b, -30c, -50b)
BHPC (0.535 g, 1.19 mmol) was dissolved in 2.20 ml of DMSO under an argon atmosphere, and a solution containing MDI (0.997 g, 3.95 mmol) in 8.00 ml of DMSO was added gradually. After the mixture was stirred at 70 °C for 10 min, a solution of PCD (2.37 g, 2.37 mmol) in 7.40 ml of DMSO was added. Then, a solution of BD (0.036 g, 0.356 mmol) in 0.50 ml of DMSO was added, and the mixture was stirred at 70 °C for 1 h. The reaction mixture was poured into excess methanol to yield a white precipitate. The precipitate was collected by filtration and purified by reprecipitation from DMSO to excess methanol. Finally, the product was dried in vacuo to afford SPUPC-30b as a yellow powder. Yield: 2.28 g (57.9%).
1H-NMR, δ (400 MHz, DMSO-d6,) (p.p.m.) (Supplementary Information): 1.28 (-OCH2CH2CH2CH2CH2CH2OCO-, m), 1.55 (-OCH2CH2CH2CH2CH2CH2OCO-, m), 1.64 (-OCH2CH2CH2CH2OCO-, m), 3.09 (-N+(CH3)3, s), 3.51 (-POCH2CH2-N, m), 3.74 (-Ph-CH2-Ph-, bs), 4.00 (-OCH2CH2OPh-, -OCH2CH2CH2CH2OCO-, -OCH2CH2CH2CH2CH2CH2OCO-, m), 4.15 (-COOCH2CH2OP-, bs), 4.24 (-POCH2CH2-N, bs), 4.39 (-OCH2CH2OPh-, -COOCH2CH2OP-, m), 6.82 (-Ph-, m), 7.07 (-Ph-, m), 7.33 (-Ph-, m), 9.45 (-NHCOO-, bs), 9.67 (-NHCOO-, bs).
IR, ν (KBr, cm−1): 3340 (N-H), 2938, 2860, 1735 (C=O), 1597, 1530, 1404, 1377, 1310 (P=O), 1240, 1217, 1174, 1061, 1018, 918, 814, 765.
SPUPC-30c and SPUPC-50b were prepared by a procedure similar to that described above, by changing the molar ratios of BHPC, PCD and BD, as shown in Table 1.
Preparation of SPU
PCD (7.00 g, 7.00 mmol) was dissolved in a flask with 7.0 ml of DMSO under an argon atmosphere, and a solution containing MDI (2.52 g, 10.0 mmol) in 10.0 ml of DMSO was gradually added. After the mixture was stirred at 70 °C for 1 h, a solution of 1,3-bis(2-hydroxyethyl)benzene (0.595 g, 3.00 mmol) was added, and the mixture was stirred at 70 °C for 25 min. The reaction mixture was poured into excess methanol to yield a white precipitate. The precipitate was collected by filtration and purified by reprecipitation from DMSO to excess methanol. Finally, the product was dried in vacuo to afford SPU as a white powder. Yield: 8.63 g (85.3%).
1H-NMR, δ (400 MHz, DMSO-d6, p.p.m.): 1.34 (-OCH2CH2CH2CH2CH2CH2OCO-, m), 1.55 (-OCH2CH2CH2CH2CH2CH2OCO-, m), 3.75 (-Ph-CH2-Ph-, bs), 4.05 (-OCH2CH2OPh-, -OCH2CH2CH2CH2CH2CH2OCO-, m), 4.18 (-OCH2CH2OPh-, m), 4.40 (-OCH2CH2O-Ph-, m), 6.55 (-Ph-, m), 7.08 (-Ph-, m), 7.34 (-Ph-, m), 9.45 (-NHCOO-, bs), 9.67 (-NHCOO-, bs).
IR, ν (KBr, cm−1): 3339 (N-H), 2940, 2864, 1705 (C=O), 1597, 1530, 1404, 1383, 1300, 1242, 1220, 1065, 1016, 914, 814, 770.
Preparations of polymer films
The synthesized polyurethanes were dissolved in DMSO, and the solution (8–10 wt %) was poured onto a Teflon or poly(ethylene terephthalate) sheet (Du Pont Japan Ltd., Tokyo, Japan). The solvent was removed at 80 °C for 3 days under its vapor atmosphere. The obtained films were then dried in vacuo at 90 °C overnight, and the self-standing films were obtained.
Characterizations
1H-NMR spectra were obtained with a JEOL NM-TH5SK 400 MHz Fourier-transform nuclear magnetic resonance spectrometer (Jeol, Tokyo, Japan) , and chemical shifts were estimated in units of p.p.m. with tetramethylsilane as an internal standard. IR spectra were recorded with a Shimadzu FTIR-8400 spectrometer (Shimadzu, Kyoto, Japan). The molecular weights of polymers were estimated with a Tosoh gel permeation chromatography (Tosoh, Tokyo, Japan) system equipped with a CCPD pump, four columns of TSKgel Multipore (Tosoh Corporation) HXL-M, a CO-8010 column oven and RI-8010 detector using DMF as an eluent. The average molecular weights were calibrated based on polystyrene standards.
Differential scanning calorimetry and thermal gravimetric analysis were performed on DSC-6200 and TG/DTA-6200 machines, respectively, from Seiko Instruments (Chiba, Japan) at a heating rate of 10 °C min−1 under a nitrogen atmosphere. The surfaces of the polymer films were analyzed with an X-ray photoelectron spectroscope (ULVAC-PHI Quantum 2000 XPS, ULVAC, Inc., Kanagawa, Japan); the take-off angle of photoelectrons was adjusted to be 90°.
Measurements of stress–strain behavior
The polymer films were cut into rectangular strips with a length of 40 mm, a width of 10 mm and a thickness of 0.15–0.30 mm. Stress–strain curves were obtained on a JT Torsi LSC-01/30; the gauge length was 20 mm and the crosshead speed was 6.0 mm min−1.
Quantitative analysis of protein adsorbed to the films
The films were cut into circular pieces with a diameter of 14 mm and immersed in phosphate-buffered solution (pH=7.4) at room temperature overnight to equilibrate the surface. The treated films were incubated for 2 h at 37 °C in phosphate-buffered solution containing protein (5 mg ml−1), anti-human albumin and fibrinogen. After 2 h, the films were removed from the solution and rinsed in excess distilled water to remove nonadsorbed proteins. The films were then immersed in a 1.0 wt % aqueous solution of SDS to remove the adsorbed proteins. The concentration of fibrinogen or albumin in the SDS solution was measured with a Micro BCATM Protein Assay kit (Thermo Fisher Scientific K.K., Kanagawa, Japan), and the number of adsorbed proteins on the membranes was estimated based on the absorbance of a protein dilution system using a Bio-Rad microplate reader (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Results and discussion
Synthesis of PC-containing diol monomer
The synthetic route of the desired diol monomer, 2-(3,5-bis(2-hydroloxyethoxy)benzoyloxy)ethyl phosphorylcholine (BHPC), is outlined in Scheme 1. First, the carboxyl compound, 4, was prepared in high yield by etherification of methyl 3,5-dihydroxybenzoate, 1, and 2-benzyloxyethyl tosylate, 2, followed by hydrolysis of the obtained ester, 3. Terminal benzyl groups were introduced into this compound as a protection group of the diol moiety. The key intermediate, 5, was synthesized by esterification of 4 with 2-bromoethanol, using K2CO3 as a base. This reaction gave 5 in good yield, but a small amount of bromoethyl ester byproduct was produced, which was obtained from esterification between the carboxyl group of 4 and the hydroxyl group of 2-bromoethanol. Thus, we have attempted to prepare 5 via the esterification of an excess of 1,2-ethanediol with the acid chloride of 4, which was obtained by reaction of 4 and thionyl chloride. Consequently, the yields of both methods were almost same (ca. 70%).
Next, the incorporation of the PC group was achieved by reaction of 5 with COP, followed by the ring-opening reaction of the cyclic phosphoric ester moiety with trimethylamine. In this reaction, purification of the intermediate, 2-[2-(3,5-bis(2-(benzyloxy)ethoxy)benzoyloxy)ethyl]-2-oxo-1,3,2-dioxaphospholane, was difficult because it was easily hydrolyzed. However, extraction of the crude products with chloroform by washing with distilled water gave the pure product. The reaction of the product with an excess of trimethylamine gave compound 6 in good yield. Finally, deprotection of the benzyl groups of 6 by Pd catalyzed hydrogen reduction with H2 gas to afford the desired diol monomer, BHPC. This reaction proceeded quantitatively to give the pure product of BHPC as a white solid, although it was so hygroscopic that the obtained solid softened when exposed to moisture.
Preparation and solubility of PC-containing polyurethane
The obtained novel diol monomer, BHPC, would be useful for the synthesis of various polymers, such as polyesters and polyurethanes, containing PC groups in the side chain. In this study, we have attempted to prepare SPUs, which were expected to be elastic biomaterials, by polyaddition using BHPC as a monomer. Before polymerization, BHPC should be completely dried in vacuo at 80 °C for a long time because it is very hygroscopic.
Two different types of SPUs were prepared, one of which consisted of two segments derived from BHPC and PCD with MDI, and the other of which consisted of three segments derived from BHPC, PCD and BD with MDI, as shown in Scheme 2. The first type of SPU was prepared by polyaddition of BHPC with excess MDI in DMSO, followed by addition of PCD to complete the polymerization. In this polymerization, the monomer ratio of BHPC and PCD was changed to obtain four copolymers (SPUPC-10a, SPUPC-20a, SPUPC-30a and SPUPC-50a), the compositions of which were different. Then, the second type of polyurethane was prepared in three steps in which BD was finally added as a chain extender, following the polyaddition reactions of BHPC with excess MDI and PCD. The compositions of the copolymers were also controlled by changing the molar ratios of BHPC, PCD and BD to obtain three polyurethanes with different compositions (SPUPC-30b, SPUPC-30c, SPUPC-50b). Furthermore, the SPU without PC units was prepared as a reference sample using PCD, BD and MDI as monomers.
The compositions and molecular weights of the obtained polyurethanes are summarized in Table 1. The chemical structures of these copolymers were confirmed by 1H-NMR and IR spectra, as described in the Experimental Procedure. The compositions of the PC units in the SPUPC series were determined from the ratio of the peak intensities of the methyl protons of the ammonium group in the PC moiety (3.09–3.10 p.p.m.) and of the methylene protons of the diphenylmethane moiety of the MDI component (3.74–3.75 p.p.m.), which existed in each monomer component. As shown in Table 1, the PC content in the copolymers could be controlled in the range of 10–50 mol%, which was in good agreement with the molar ratio of BHPC used in the polymerization. The mass contents (wt%) of the PC units in SPUPC-30b, SPUPC-30c and SPUPC-50b were higher than those of SPUPC-30a and SPUPC-50a, respectively, although the molar content (mol%) of the copolymers was nearly the same. The introduction of butylene moieties instead of a polycarbonate moiety increased the mass content of the PC units in the copolymers, because the molecular weight of BD was smaller than that of PCD. The Mn of the obtained polymers was over 106, which were estimated by gel permeation chromatography using DMF as the eluent. These values must be higher than the real molecular weights; it was assumed that the molecular chain of these copolymers would be fully extended in DMF and would result in a very large free volume of molecules in such a polar solvent, caused by the high polarity of the PC moiety in the side chain. We speculate that the real values of Mn of the SPUPC series would be on the same order of magnitude as the Mn values of SPU listed in Table 1. Moreover, high molecular weight polyurethanes could be prepared using BHPC as a diol monomer.
Table 2 lists the solubility of the obtained copolymers. SPU, SPUPC-10a, SPUPC-20a and SPUPC-30a exhibited good solubility in chloroform, THF and aprotic polar solvents, such as N,N-dimethylacetamide, DMF and DMSO, but were insoluble in ethanol and water. Both SPUPC-30b and SPUPC-30c, which contained butylene units derived from BD in the main chain, were soluble in aprotic polar solvents and partially soluble in chloroform and THF. Their solubility in specific solvents is advantageous for processing medical devices, and their insolubility in other solvents allows the material to be durable in these solvents. However, SPUPC-50a and SPUPC-50b were insoluble in water, ethanol, chloroform and THF, and were partially soluble even in aprotic polar solvents. Therefore, the solubility of these copolymers decreased with increasing PC content; the maximum PC content in these copolymers that allowed for solubility in aprotic polar solvents was ca. 30 wt%. It was speculated that a polar PC group in the side chain would have a strong interaction with the polar urethane unit in the main chain, which would make the polymer insoluble even in aprotic polar solvents.
Physical properties of PC-containing polyurethane
The thermal properties of these copolymers were investigated by differential scanning calorimetry and thermogravimetric analysis. In the differential scanning calorimetry thermograms, the glass transition temperature and the melting temperature were not observed in the range between −100 °C and 250 °C for every polymer in the SPUPC series, which suggests that the glass transition temperature of these polyurethanes was >250 °C. From thermal gravimetric analysis measurements, it was found that weight loss of the SPUPC series started at ca. 250 °C. The weight loss was initiated by thermal degradation of the polymer side chain, which contained PC groups or spacer moieties. Therefore, the heat resistance of these PC-containing polyurethanes until 250 °C would be sufficient for use in medical devices, for example, for thermal sterilization processes above 150 °C.
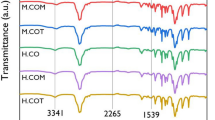
The surface structures of films of the SPUPC series were analyzed by X-ray photoelectron spectroscopy. The X-ray photoelectron spectroscopy spectra of SPUPC-30a and SPUPC-30c are shown in Figure 1. The peaks of N1s and P2p were observed at 402.5 and 133.0 eV, respectively, which were attributed to the nitrogen atom of the ammonium group and to the phosphorus atom of the phosphate group in the PC moiety, respectively. In addition, the static contact angle of water on the film surface of SPUPC-30a was observed at 73°, whereas that of the SPU film was 75°. Therefore, it was suggested that the PC moiety exists to some extent on the film surfaces of the SPUPC series, although the peak intensity of the PC moiety in the X-ray photoelectron spectroscopy spectra was not strong, and the contact angle of water in PC-containing polyurethane films was almost same as that in the SPU film. These results demonstrate that PC-containing polyurethanes possess similar thermal and surface properties to PC-containing polyamides and poly(urethane-urea)s, the properties of which were reported in our previous papers.22, 23, 24, 25

X-ray photoelectron spectroscopy (XPS) spectra of the films of (a) SPUPC-30a and (b) SPUPC-30c. A full color version of this figure is available at Polymer Journal online.
The stress–strain behavior of SPUPC films was evaluated to reveal the effect of PC content on mechanical properties. For these experiments, SPUPC-10a, SPUPC-20a, SPUPC-30a, SPUPC-30b and SPUPC-30c were used as film samples. The homogeneous films of SPUPC-50a and SPUPC-50b could not be obtained because they were partially soluble in DMSO, and the insoluble residue remained in solution and made the films brittle. Figure 2 shows the stress–strain behavior of the copolymer films; the Young’s modulus, the tensile strength and the elongation to break are summarized in Table 3.

Stress–strain behavior of the films of PC-containing polyurethanes.
As shown in Figure 2, these copolymer films underwent a large elongation from 200 to 800%, which appeared to have rubber-like elasticity. In particular, a large elongation was observed for films of SPUPC-10a and SPUPC-20a, which could be due to the high content of soft segments of polycarbonate units, above 85 wt%. This stress–strain behavior was similar to that of polyurethanes containing polycarbonate as the soft segment, which was previously described in the literature.28, 29, 30, 31 However, the films of SPUPC-30a, SPUPC-30b and SPUPC-30c possessed higher Young’s moduli than those of SPUPC-10a and SPUPC-20a. Therefore, the PC-containing monomer unit acted as a hard segment in the copolymer films, and its Young’s modulus increased with increasing incorporation of PC groups. In addition, the introduction of a butylene unit in the main chain induced higher tensile strength in the films. The effects of PC content and composition of the copolymers on the mechanical properties are in good agreement with our previous results24 of poly(urethane-urea)s containing PC moieties. Among these copolymer films, the film of SPUPC-30a exhibited the best performance of elastic properties, which had a high Young’s modulus and tensile strength in addition to a relatively high PC content.
Protein adsorption properties of PC-containing polyurethane films
For preliminary evaluation of biocompatibility, the concentration of fibrinogen and albumin on PC-containing polyurethane films was estimated after the films were immersed in the protein solutions for 2 h, as shown in Figure 3. The films of poly(ethylene terephthalate), SPU and MPC polymer, poly(MPC-co-butyl methacrylate; MPC content: 30 mol%) were used as reference samples, and the protein adhesion to the cast films of SPUPC-10a, SPUPC-20a, SPUPC-30a, SPUPC-30b and SPUPC-30c were compared with that to the films of the reference samples. The MPC polymer film was prepared by coating of a poly(ethylene terephthalate) substrate with an ethanol solution of MPC polymer.

Amount of adherent fibrinogen and albumin on the film surfaces after incubation in these protein solutions in phosphate-buffered solution for 2 h at 37 °C. Each bar represents the mean±s.e. of four experiments.
As shown in Figure 3, protein adsorption was significantly suppressed on the film surfaces of the SPUPC series compared with poly(ethylene terephthalate) and SPU owing to the additive effect of PC moieties to the potential biocompatibility of SPU. The amount of protein adsorbed decreased with increasing PC content in these copolymers. In particular, the amounts of adsorbed fibrinogen and albumin on the SPUPC-30c film were 0.412 and 0.355 μg cm−2, respectively, whereas those on the SPU film were 1.04 and 0.702 μg cm−2, respectively. In addition, the amount of adsorbed proteins on SPUPC-30c was the same as that on the MPC polymer. Therefore, a PC content of ca. 30 wt % would be sufficient to reduce protein adsorption to these PC-containing polyurethanes, and the composition of the PC moiety would be a dominant factor in reduction of protein adsorption.
Conclusion
A novel diol compound containing a PC moiety, BHPC, was successfully synthesized, and several types of SPUs were prepared from BHPC with a polycarbonate-based soft segment. The polyurethane films obtained exhibited thermal stability up to 250 °C and elastic mechanical properties; the Young’s modulus increased with increasing PC content up to 31 wt%. The introduction of such a polar phospholipid group was effective in improving the resistance to protein adhesion of the polymer film, which was the result of surface properties derived from the PC moiety. Therefore, a thermally stable and highly elastic polyurethane material containing PC moieties was obtained in this study. Consequently, it is expected that such polyurethanes will be useful as polymeric materials to develop new biomedical devices, although more detailed biochemical assays and clinical research must be performed. In addition, it was also found that there was a trade-off between the solubility and the PC content of polyurethanes. To obtain a soluble polymer with high PC content, a different concept for the molecular design or the polymerization process of polymers containing PC moieties should be developed, which is currently in progress.

Synthesis of a diol monomer containing a PC moiety.

Preparations of segmented polyurethane containing PC (SPUPC) moieties.
References
Lelah, M. D. & Cooper, S. L. Polyurethanes in Medicine, (CRC Press, Boca Raton, 1986).
Eisenbach, C. D., Fischer, K., Hayen, H., Nefzger, H., Ribbe, A. & Stadler, E. Polymeric Materials Encyclopedia Vol. 9, pp 6957–6968 (CRC Press, Boca Raton, 1996).
Zhao, Q., Topham, N., Anderson, J. M., Hiltner, A., London, G. M. & Payet, C. R. Foreign-body giant cells and polyurethane biostability: In vivo correlation of cell adhesion and surface cracking. J. Biomed. Mater. Res. 25, 177–183 (1991).
Wu, Y., Zhao, Q., Anderson, J. M., Hiltner, A., London, G. M. & Payet, C. R. Effect of some additives on the biostability of a poly(ether urethane) elastomer. J. Biomed. Mater. Res. 25, 725–798 (1991).
Zhao, Q. H., McNally, A. K., Rubin, K. R., Renier, M. & Wu, Y. V. Human plasma α2-macroglobulin promotes in vitro oxidative stress cracking of pellethane 2363-80A: In vivo and in vitro correlations. J. Biomed. Mater. Res. 27, 379–388 (1993).
Stokes, K., McVenes, R. & Anderson, J. M. Polyurethane Elastomer Biostability. J. Biomater. Appl. 9, 321–354 (1995).
Kang, I. K., Kwon, O. H., Kim, M. K., Lee, Y. M. & Sung, Y. K. In vitro blood compatibility of functional group-grafted and heparin-immobilized polyurethanes prepared by plasma glow discharge. Biomaterials 18, 1099–1107 (1997).
Mathur, A. B., Collier, T. O., Kao, W. J., Wiggins, M., Schubert, M. A., Hiltner, A. & Anderson, J. M. In vivo biocompatibility and biostability of modified polyurethanes. J. Biomed. Mater. Res. 36, 246–257 (1997).
Flemming, R. G., Proctor, R. A. & Cooper, S. L. Bacterial adhesion to functionalized polyurethanes. J. Biomater. Sci. Polym. Ed. 10, 679–697 (1999).
Roh, H. W., Song, M. J., Han, D. K., Lee, D. S., Ahn, J. H. & Kim, S. C. Effect of cross-link density and hydrophilicity of PU on blood compatibility of hydrophobic PS/hydrophilic PU IPNs. J. Biomater. Sci. Polym. Ed. 10, 123–143 (1999).
Lee, J. H., Ju, Y. M. & Kim, D. M. Platelet adhesion onto segmented polyurethane film surfaces modified by addition and crosslinking of PEO-containing block copolymers. Biomaterials 21, 683–691 (2000).
Khan, I., Smith, N., Jones, E., Finch, D. S. & Cameron, R. E. Analysis and evaluation of a biomedical polycarbonate urethane tested in an in vitro study and an ovine arthroplasty model. Part I: materials selection and evaluation. Biomaterials 26, 621–631 (2005).
Khan, I., Smith, N., Jones, E., Finch, D. S. & Cameron, R. E. Analysis and evaluation of a biomedical polycarbonate urethane tested in an in vitro study and an ovine arthroplasty model. Part II: in vivo investigation. Biomaterials 26, 633–643 (2005).
Ishihara, K., Ueda, T. & Nakabayashi, N. Preparation of phospholipid polymers and their properties as polymer hydrogel membranes. Polym. J. 22, 355–360 (1990).
Ueda, T., Oshida, H., Kurita, K., Ishihara, K. & Nakabayashi, N. Preparation of 2-methacryloxyethyl phosphorylcholine copolymers with alkyl methacrylates and their blood compatibility. Polym. J. 24, 1259–1269 (1992).
Ishihara, K., Hanyuda, H. & Nakabayashi, N. Synthesis of phospholipid polymers having a urethane bond in the side chain as coating material on segmented polyurethane and their platelet adhesion-resistant properties. Biomaterials 16, 873–879 (1995).
Ishihara, K., Tanaka, S., Furukawa, N., Kurita, K. & Nakabayashi, N. Improved blood compatibility of segmented polyurethanes by polymeric additives having phospholipid polar groups I. Molecular design of polymeric additives and their functions. J. Biomed. Mater. Res. 32, 391–399 (1996).
Ishihara, K., Shibata, N., Tanaka, S., Iwasaki, Y., Kurosaki, T. & Nakabayashi, N. Improved blood compatibility of segmented polyurethane by polymeric additives having phospholipid polar group II. Dispersion state of the polymeric additive and protein adsorption on the surface. J. Biomed. Mater. Res. 32, 401–408 (1996).
Iwasaki, Y., Aiba, Y., Morimoto, N., Nakabayashi, N. & Ishihara, K. Semi-interpenetrating polymer networks composed of biocompatible phospholipid polymer and segmented polyurethane. J. Biomed. Mater. Res. 52, 701–708 (2000).
Ishihara, K. & Iwasaki, Y. Biocompatible elastomers composed of segmented polyurethane and 2-methacryloyloxyethyl phosphorylcholine polymer. Polym. Adv. Technol. 11, 626–634 (2000).
Morimoto, N., Iwasaki, Y., Nakabayashi, N. & Ishihara, K. Physical properties and blood compatibility of surface-modified segmented polyurethane by semi-interpenetrating polymer networks with a phospholipid polymer. Biomaterials 23, 4881–4887 (2002).
Nagase, Y., Oku, M., Iwasaki, Y. & Ishihara, Y. Preparations of aromatic diamine monomers and copolyamides containing phosphorylcholine moiety and the biocompatibility of copolyamides. Polym. J. 39, 712–721 (2007).
Horiguchi, K., Shimoyamada, N., Nagawa, D., Nagase, Y., Iwasaki, Y. & Ishihara, K. Syntheses of a novel diamine monomer and aromatic polyamides containing phosphorylcholine group. Trans. Mater. Res. Soc. Jpn 33, 1261–1264 (2008).
Nagase, Y., Nakajima, S., Oku, M., Iwasaki, Y. & Ishihara, K. Synthesis and properties of segmented poly(urethane-urea)s containing phosphorylcholine moiety in the side-chain. Polym. J. 40, 1149–1156 (2008).
Nagase, Y. & Horiguchi, K. Biocompatible Polyamides And Polyurethanes Containing Phospholipid Moiety pp 217–232 (Biomedical Engineering—Frontiers and Challenges, InTech, Croatia, 2011).
Yung, L. L. & Cooper, S. L. Neutrophil adhesion on phosphorylcholine-containing polyurethane. Biomaterials 19, 31–40 (1998).
Edmundson, R. S. Oxidation of cyclic phosphorochloridites. Chem. Ind. 1828–1829 (1962).
Tanaka, H. & Kunimura, M. Mechanical properties of thermoplastic polyurethanes containing aliphatic polycarbonate soft segments with different chemical structures. Polym. Eng. Sci. 42, 1333–1349 (2002).
Eceiza, A., Martin, M. D., Caba, K., de la, Kortaberria, G., Gabilondo, N., Corcuera, M. A. & Mondragon, I. Thermoplastic polyurethane elastomers based on polycarbonate diols with different soft segment molecular weight and chemical structure: mechanical and thermal properties. Polym. Eng. Sci. 48, 297–306 (2008).
Kojio, K., Furukawa, M., Motokucho, S., Shimada, M. & Sakai, M. Structure-mechanical property relationships for poly(carbonate urethane) elastomers with novel soft segments. Macromolecules 42, 8322–8327 (2009).
Chung, Y. C., Choi, J. W., Shin, S. J. & Chun, B. C. Laterally crosslinked polyurethane copolymers with polycarbonate as a soft segment. Fiber Polym 13, 815–822 (2012).
Acknowledgements
We express our sincere gratitude to Professor Kazuhiko Ishihara and Professor Yasuhiko Iwasaki, who are from the School of Engineering, University of Tokyo and from the Faculty of Chemistry, Materials and Bioengineering, Kansai University, respectively, for their scientific and technical support. We also thank Professor Takashi Asaka from the Department of Applied Chemistry, Tokai University, for his help in conducting the stress–strain measurements. This work was partially supported by a Grant-in-Aid for Research from the Tokai University Support Association Union.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Polymer Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Sakagami, Y., Horiguchi, K., Narita, Y. et al. Syntheses of a novel diol monomer and polyurethane elastomers containing phospholipid moieties. Polym J 45, 1159–1166 (2013). https://doi.org/10.1038/pj.2013.48
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pj.2013.48
