
Abstract
A series of novel fluorene moiety-containing polycarbonates was synthesized by the anionic ring-opening polymerization of six-membered spirocyclic carbonates possessing 9,9-fluorene groups at the 5,5-position. The polymers could be considered cardo-type polymers because they had 9,9-fluorene groups as a loop-shaped side chain. Interestingly, the fluorene pendant polycarbonates were obtained in sufficiently high molecular weights and in good yields, although the monomers were structurally closely related to the 5,5-diphenyl-substituted cyclic carbonate that exhibited very low polymerizability. Furthermore, the polymerizations of substituted fluorene-based six-membered spirocyclic carbonates were investigated. All of the obtained polymers showed favorable properties, including good solubility, amorphous natures, favorable thermal stabilities, remarkably low birefringence values and high refractive indices despite consisting only of C, H and O atoms. The good potentiality in the application to optical materials was shown here.
Similar content being viewed by others
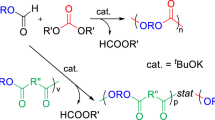

Introduction
Polymers containing 9,9-diarylfluorene moieties in the main chain, the so-called ‘cardo polymers’, have attracted considerable attention in a variety of fields because of their high performance and good optical properties, including high refractive index, low birefringence and high transparency. Furthermore, some of them have good processability because of their appropriate glass transition and high decomposition temperatures, as well as good solubility in organic solvents.1, 2 We have long studied the synthesis and properties of many cardo polymers having 9,9-diarylfluorene moieties3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16 and recently reported the preparation of cardo-like polymers17, 18, 19, 20, 21 that have 9,9′-spirobifluorene moieties as the main chain units. Most cardo polymers hitherto studied have been aromatic, whereas aliphatic cardo polymers, ‘cardo carbochain polymers’ as defined by Korshak,1 are also interesting because of their expected moderate properties based on their flexible main chains, although only a few reports dealing with fluorene-containing polymers or oligomers have been reported so far, to the best of our knowledge.22, 23, 24, 25, 26, 27 Nakano et al.25, 26, 27 reported the preparation of polydibenzofulvenes with interesting properties directed toward possible applications in optical materials. Among the cardo carbochain polymers, polycarbonates seem of particular interest because of their excellent properties such as high transparency and thermal stability as well as biodegradability. We have synthesized a few aliphatic polycarbonates with 9,9′-spirobifluorene moieties as one of the cardo-like aliphatic polymers via polycondensation.19 Meanwhile, ring-opening polymerization (ROP) of six- or seven-membered cyclic carbonate28, 29, 30 is one of the typical synthetic entries for polycarbonates without any by-product formation. However, two bulky substituents at the five-position of six-membered cyclic carbonate, such as phenyl groups, mostly diminish its polymerizability because of the equilibrium polymerization, as reported by Endo et al.31, 32 Polycarbonates derived by ROP have an additional merit from another point of view: Takata and Endo reported the volume expansion during the ROP of cyclic carbonates33, 34, 35, 36 and their sulfur analogs,37 suggesting the possible control of their internal stress and optical distortion in actual use. Furthermore, ROP can be carried out by versatile methods such as cationic,36, 38, 39 anionic,31, 40 coordination-insertion41, 42, 43 and enzyme-catalyzed44, 45, 46 polymerizations. We have studied the anionic ROP of six-membered cyclic carbonates, substituted spiro[fluorene-9,5′-[1,3]-dioxan]-2′-ones and have obtained corresponding aliphatic polycarbonates with fluorene groups as a new class of cardo carbochain polymers with interesting properties.47 Recently, Hedrick’s report on a copolymer involving a fluorene-tethered polycarbonate as a copolymer unit has prompted us to report our results on the synthesis and properties of aliphatic polycarbonates having substituted and unsubstituted fluorene moieties in the main chain.48 Herein, we describe the synthesis of the aliphatic polycarbonates by anionic ROP of fluorene-containing six-membered spirocyclic carbonate monomers. The properties of the resulting polymers and the volume change during the polymerization are also discussed.
Experimental procedure
Materials and methods
Monomers 3-7 were prepared from 2,7-, or 3,6-dibromofluorene via Suzuki-Miyaura coupling reaction, hydroxymethylation and cyclization to spirocyclic carbonates according to typical methods (see supporting information). The other materials were commercially available and were used without further purification. The polymerization was conducted under an argon atmosphere in dry tetrahydrofuran (THF) or N,N-dimethylformamide (DMF). THF was dried over sodium and distilled under a nitrogen atmosphere before use. DMF was dried over CaH2 and distilled under reduced pressure.
Characterization
The melting points were measured on a SMP3 melting point apparatus (Stuart Scientific, Staffordshire, UK). 1H and 13C NMR spectra were recorded on a JEOL AL-400 NMR spectrometer (JEOL, Tokyo, Japan) operating at 400 and 100 MHz, respectively, in CDCl3 or dimethyl sulfoxide-d6 and with tetramethylsilane as an internal standard. IR spectra were recorded on a JASCO FT/IR-460 plus spectrometer (JASCO, Tokyo, Japan). The molecular weights (Mn and Mw) and polydispersity (Mw/Mn) of the polymers were measured using size exclusion chromatography at 30 °C with DMF (10 mM LiBr, 0.85 ml min–1) on a JASCO HSS-1500 system equipped with consecutive linear polystyrene gel columns (TOHSO TSK gel G2000HXL and GMHXL) and were calculated based on a polystyrene calibration. High-resolution mass spectra FAB were obtained with a JEOL JMS-700 at the Center for Advanced Materials Analysis, Tokyo Institute of Technology, Tokyo, Japan. Thermogravimetric analyses were carried out with a Shimadzu TGA-50 (heating rate of 10 °C min–1) under the use of nitrogen. Differential scanning calorimetry was performed using a Shimadzu DSC-60 instrument under nitrogen (heating rate of 10 °C min–1). The refractive indices and retardations of poly 2 were measured using a Kalnew precision refractometer (KPR-30, Shimadzu, Kyoto, Japan).
Synthesis of 9,9-bis(hydroxymethyl)fluorene 1
Fluorene (30.0 g, 180 mmol) and paraformaldehyde (43.3 g, 1.44 mol) were dissolved in a mixed solvent of dimethyl sulfoxide (180 ml) and MeOH (10 ml), and the mixture was degassed. NaOMe (5.0 M in MeOH, 145 ml, 722 mmol) was added to the mixture from a dropping funnel at 0 °C under an argon atmosphere, and then the resulting mixture was stirred at 0 °C for 1 h. The reaction mixture was acidified with concentrated HCl, diluted with water (200 ml), and extracted with ethyl acetate (100 ml × 3). The combined organic layer was washed with water, dried over anhydrous MgSO4, filtered and concentrated under reduced pressure. The residual product was purified by recrystallization from toluene to give 9,9-bis(hydroxymethyl)fluorene 1 (24.6 g, 112 mmol, 62%) as a white needle, mp. 146.0–146.9 °C (Lit.50 mp. 145.5–146.5 °C); 1H NMR (400 MHz, CDCl3, 298 K) δ 7.77 (d, J=7.6 Hz, 4H), 7.61 (d, J=7.5 Hz, 2H), 7.42 (t, J=7.5 Hz, 2H), 7.33 (t, J=7.5 Hz, 2H), 4.00 (s, 4H), 2.19 (t, 2H) p.p.m.; 13C NMR (100 MHz, CDCl3, 298 K) δ 145.75, 140.97, 128.13, 127.13, 124.49, 120.26, 66.81, 57.31 p.p.m.; IR (KBr) ν 3313(br), 2973, 2941, 2872, 1476, 1447, 1376, 1066, 1041, 1024, 994, 761, 755, 559 cm–1.
Synthesis of spiro[fluorene-9,5′-[1,3]-dioxan]-2′-one 2
Ethyl chloroformate (9.9 ml, 103 mmol) was added to a solution of 1 (11.6 g, 51.4 mmol) in dry THF (130 ml). Triethylamine (14.3 ml, 103 mmol) was slowly added dropwise into the mixture at 0 °C. After stirring for 30 min at 0 °C, the mixture was warmed up to room temperature and stirred for an additional 2 h. The precipitates formed were removed by filtration, and the filtrate was washed with aq. NH4Cl, dried over MgSO4, filtered and concentrated. The residual product was purified by recrystallization from toluene to give 2 (8.02 g, 56.7 mmol, 55%) as a white solid mp. 156.5 °C (decomp.); 1H NMR (400 MHz, CDCl3, 298 K) δ 7.81 (d, J=8.0 Hz, 2H), 7.53–7.49 (m, 4H), 7.39 (t, J=7.5 Hz, 2H), 4.59 (s, 4H) p.p.m.; 13C NMR (100 MHz, CDCl3, 298 K) δ 148.20, 142.45, 140.56, 129.36, 128.14, 124.01, 120.70, 74.87, 46.70 p.p.m.; IR (KBr) ν 3048, 2994, 2972, 2902, 1930, 1762, 1479, 1458, 1406, 1258, 1196, 1162, 1138, 1108, 760, 734, 622, 591, 574 cm–1; high-resolution mass spectra (FAB) m/z Calcd for C16H12O3 [M]+=252.0786, found 252.0790.
Typical polymerization procedure
All glass vessels used were heated in vacuo before use, filled with dry argon and handled under an argon atmosphere. To a suspension of cyclic carbonates in dry THF in a dried Schlenk tube equipped with a magnetic stir bar, initiator was added at a set temperature to start the polymerization. The reaction was quenched with the addition of phosphoric acid (10 vol% methanol solution, 2 ml) to the reaction mixture, and the mixture was purified by reprecipitation from methanol (100 ml) to give the corresponding polycarbonates as the precipitates.
Polymerization of 2
Monomer 2 (0.500 g, 1.98 mmol) was suspended in THF (2.0 ml), and 0.020 mmol of initiator was added (Run 1: t-BuOK (1.0 M in THF, 20 μl), Run 2-3: n-BuLi (2.6 M in hexane, 7.7 μl), Run 4: s-BuLi (1.02 M in hexane, 20 μl)). After reprecipitation from methanol, poly 2 was obtained as a white solid. 1H NMR (400 MHz, CDCl3, 328 K) δ 7.65 (d, J=7.6 Hz, 2H), 7.45 (d, J=7.6 Hz, 2H), 7.31 (t, J=7.4 Hz, 2H), 7.19 (t, J=7.6 Hz, 2H), 4.32 (s, 4H) p.p.m.; 13C NMR (100 MHz, CDCl3, 328 K) δ 154.49, 143.87, 140.88, 128.71, 127.55, 125.18, 120.31, 69.15, 53.41 p.p.m.; IR (KBr) ν 3436, 3064, 2952, 2895, 1751, 1449, 1338, 1231, 968, 735 cm–1; Tg: 129 °C, Td5: 303 °C.
Polymerization of spiro[2,7-diphenylfluorene-9,5′-[1,3]-dioxan]-2′-one 3
Monomer 3 (0.100 g, 0.247 mmol) was suspended in dry DMF (0.25 ml), and s-BuLi (1.1 M in hexane, 21 μl, 23 μmol) was added at −20 °C. The procedure was carried out in the same manner as that of poly 2. However, polymers and oligomers were obtained, and the monomer was recovered.
Polymerization of spiro[2,7-bis(p-fluorophenyl)fluorene-9,5′-[1,3]-dioxan]-2′-one 4
Monomer 4 (0.100 g, 0.247 mmol) was suspended in dry DMF (0.25 ml), and s-BuLi (1.1 M in hexane, 21 μl, 23 μmol) was added at −20 °C. The procedure was carried out in the same manner as that of poly 2. However, polymers and oligomers were not obtained, and the monomer 4 was recovered.
Polymerization of spiro[3,6-diphenylfluorene-9,5′-[1,3]-dioxan]-2′-one 5
Monomer 5 (0.150 g, 0.494 mmol) was suspended in dry DMF (0.35 ml), and t-BuOK (5.5 mg, 49 μmol) or s-BuLi (0.75 M in cyclohexane, 53 μl, 49 μmol) was added at −20 or 0 °C. The procedure was carried out in the same manner as that of poly 2 to give poly 5 (entry 5: 0.126 g, entry 6: 0.102 g) as a white solid insoluble in methanol. 1H NMR (400 MHz, CDCl3, 298 K) δ 7.97–7.78 (m, 2H), 7.65–7.20 (m, 14H), 4.29 (s, 4H) p.p.m.; 13C NMR (100 MHz, CDCl3, 298 K) δ 154.64, 143.05, 141.94, 141.19, 140.88, 128.93, 127.60, 127.39, 126.92, 125.55, 119.04, 69.11, 52.67 p.p.m.; IR (NaCl) ν 3056, 3031, 2954, 2893, 1753, 1478, 1401, 1243, 967, 909, 836, 766, 733, 697 cm−1; Tg: 156 °C, Td5: 287 °C.
Polymerization of spiro[3,6-bis(p-fluorophenyl)fluorene-9,5′-[1,3]-dioxan]-2′-one 6
Monomer 6 (0.150 g, 0.341 mmol) was suspended in dry DMF (0.34 ml), and t-BuOK (3.8 mg, 34 μmol, entry 7) or s-BuLi (0.75 M in cyclohexane, 46 μl, 34 μmol, entry 8 & 9) was added at −20 or 0 °C. The procedure was carried out in the same manner as that of poly 2 to give poly 6 (entry 7: 0.123 g, entry 8: 0.117 g) as a white solid insoluble in methanol. In entry 9, the polymer could not be isolated from the mixture with oligomers. 1H NMR (400 MHz, dimethyl sulfoxide-d6, 373 K) δ 8.03 (br, 2H), 7.48 (br, 4H), 7.36–7.20 (m, 4H), 7.00 (br, 4H), 4.21 (s, 4H) p.p.m.; 13C NMR (100 MHz, CDCl3, 298 K) δ 162.69 (d, J=247 Hz), 154.50, 142.95, 141.18, 141.07, 136.83, 128.84, 126.75, 125.40, 118.91, 115.84 (d, J=21 Hz), 69.05, 52.65 p.p.m.; IR (NaCl) ν 3038, 2948, 1747, 1600, 1513, 1485, 1383, 1223, 1159, 963, 912, 843, 820, 774, 730, 622, 525 cm−1; Tg: 185 °C, Td5: 272 °C.
Polymerization of spiro[3,6-bis(p-methoxyphenyl)fluorene-9,5′-[1,3]-dioxan]-2′-one 7
Monomer 7 (0.150 g, 0.323 mmol) was suspended in dry DMF (0.43 ml), and t-BuOK (3.6 mg, 32 μmol) was added at −20 or 0 °C. The procedure was carried out in the same manner as that of poly 2 to give poly 7 (entry 10: 48 mg, entry 11: 60 mg) as a white solid insoluble in methanol. 1H NMR (400 MHz, CDCl3, 333 K) δ 7.90–7.72 (m, 2H), 7.61–7.40 (m, 8H), 6.96–6.78 (m, 4H), 4.31 (br, 4H), 3.80 (br, 6H) p.p.m.; 13C NMR (100 MHz, CDCl3, 298 K) δ 159.24, 154.55, 143.64, 142.39, 141.04, 133.34, 128.26, 126.32, 125.37, 118.52, 114.21, 68.96, 55.29, 52.53 p.p.m.; IR (NaCl) ν 3029, 2998, 2954, 2934, 2899, 2832, 1751, 1608, 1518, 1486, 1245, 1179, 1038, 822 cm−1; Tg: 119 °C, Td5: 270 °C.
Results and Discussion
Synthesis and polymerization of cyclic carbonate 2
Polymerization of the fluorene-tethering monomer (spiro[fluorene-9,5′-[1,3]-dioxan]-2′-one, 2) was studied in detail. Monomer 2 was synthesized by a two-step reaction starting from fluorene (Scheme 1). 9,9-Bis(hydroxymethyl)fluorene, 1, was prepared by the hydroxymethylation of fluorene with paraformaldehyde and NaOMe.49 Subsequent cyclization of 1 was achieved by the reaction with ethyl chloroformate according to our previously reported procedure,36 providing the corresponding cyclic carbonate 2 in 55% yield. The cyclization of 1 to 2 was confirmed by the reasonable 1H NMR spectral change. The large downfield shift of the methylene signal (Hb) from 4.00 to 4.59 p.p.m. was indicative of the electron-withdrawing effect of the carbonate skeleton and the ring current effect of the fluorene moiety. Furthermore, the 1,8-protons (Hc) of fluorene were shifted upfield by the cyclization (Figure 1(1)-a and -b). The stretching vibration signal of the carbonate carbonyl bond was observed at 1762 cm–1 in the IR spectrum, which was consistent with the typical cyclic carbonate value.

(1) 1H NMR spectra of (a) 1, (b) 2 and (c) poly 2 (400 MHz, CDCl3). (2) Calculated structures of (a) cyclic carbonate 2 and (b) 5,5-diphenyl-1,3-dioxan-2-one calculated at the B3LYP/6-31G(d.p) level. A full color version of this figure is available at Polymer Journal online.
Anionic ROP of 2 was carried out in THF with a few appropriate initiators (Scheme 1 and Table 1).
First, t-BuOK was used in accordance with the literature procedure31 at 0 °C. The initial heterogeneous solution of 2 in THF at 0 °C became homogeneous, indicating the initiation of the polymerization by t-BuOK. The time-molecular weight (Mn) relationship by size exclusion chromatography supported the formation of polymer (Table 1, Run 1). However, the Mn and dispersity hardly changed even when the reaction time was prolonged up to 120 min. These results indicated the good polymerizability of 2 along with the negligible backbiting reaction under the conditions, which was enough to give the high molecular weight of the polymer. The resultant polymer was purified by reprecipitation with methanol to give poly 2 in 84% yield. The chemical structure of poly 2 was fully characterized by the spectral data. A characteristic C=O stretching vibration was clearly observed at 1751 cm–1 in the IR spectrum, which was consistent with that of linear carbonate. In the 1H NMR spectrum, signals assignable to the methylene moiety (Hb) was shifted upfield from 4.59 to 4.32 p.p.m. because of the cancellation of the ring current effect of the fluorene moiety by the ring opening (Figure 1(1)-c and (2)-a). Other signals coincided with the structure of poly 2 to rule out the partial elimination of carbon dioxide during the polymerization. To increase the polymer molecular weight, we used the polymerization at lower temperature with t-BuOK; however, the polymer yield significantly decreased to 25%. As monomer 2 was recovered in the methanol soluble part along with a small amount of oligomers, it was considered that the polymerization could not proceed sufficiently at −20 °C because of the low solubility of 2 and poly 2. Next, n-BuLi was used instead of t-BuOK as a homogeneous initiator (Run 2).40 As expected, the polymerization of 2 proceeded quickly, whereas the prolongation of the reaction time led to a gradual decrease in the molecular weight, likely because of the emerging backbiting reaction to oligomers. When the reaction time was shortened to 5 min, a higher molecular weight poly 2 was obtained in an excellent yield (Run 3). Furthermore, s-BuLi was used because it is usually more effective than n-BuLi because of its lower association nature, which can lead to higher initiation efficiency.50, 51, 52, 53, 54 Indeed, s-BuLi resulted in the formation of a higher molecular weight poly 2 in an excellent yield (Run 4). In the time-conversion plot for the s-BuLi-initiated polymerization of 2 at −20 to 20 °C using 1H NMR (Supplementary Figures S1–S5 (see Supplementary Information)), only the disappearance of 2 and the formation of the oligomers and poly 2 were observed. At higher temperature, the signals attributable to the oligomers increased according to the decrease of 2. Based on these results, the short reaction time and low temperature effectively afforded both sufficiently high yield and high molecular weight of poly 2 by reducing the oligomerization via the backbiting reaction. Meanwhile, the fact that the monomer 2 showed good polymerizability in anionic ROP was interesting in the light of the low polymerizability of 5,5-diphenyl-1,3-dioxan-2-one, which had a similar structure to 2, even when the difference in polymerization conditions was considered.31, 32 A plausible explanation for this significant difference would come from the higher ring strain of 2 because of the spirocyclic structure or the slower backbiting reaction than 5,5-diphenyl-1,3-dioxan-2-one. As the steric hindrance around the carbonate carbon as the reaction center seemed similar in these monomers, the internal angle of six-membered ring may have an important role in the polymerizability (2, 105.3°; 5,5-diphenyl-1,3-dioxane-2-one, 103.3°), as shown in their calculated structures (Figure 1(2)). In fact, the above NMR study indicated the absence of 2 in the equilibrium stage, which was clearly different from the case of 5,5-diphenyl-1,3-dioxan-2-one,31, 32 although the oligomer formation was confirmed (see Supplementary Information).
One of the most interesting properties of the ROP of cyclic carbonate results in the occurrence of volume expansion upon polymerization from monomer to polycarbonate,33, 34, 35, 36 although most monomers shrink in volume during polymerization. To evaluate the degree of volume expansion associated with the ROP of cyclic carbonate, the densities of 2 and poly 2 were measured. The density of 2 and poly 2 were 1.3667 and 1.3202 g cm–3, respectively. These results revealed the certain volume expansion of 3.5%, which was consistent with values previously reported for similar ROPs of six-membered cyclic carbonates.33, 34, 35, 36
Synthesis and polymerization of substituted derivatives of 2
The substituent effect on the fluorene moiety was examined using several additional monomers 3-7, that is, 2,7- and 3,6-substituented derivatives of 2 (Table 2). These monomers were synthesized by the Suzuki-Miyaura coupling reactions of 2,7- or 3,6-dibromofluorenones and the corresponding arylboronic acids, followed by the reduction, hydroxymethylation and carbonate-cyclization via similar procedures mentioned above.50 Subsequent anionic ROP of 3-7 (from −20 to 0 °C) was carried out in DMF because of their poor solubility. The 2,7-disubstituted monomers 3 and 4, in particular, were hardly soluble even in DMF at 0 °C. Consequently, no polymer was obtained by this method, and only monomers and a trace amount polymer were recovered, as detected by size exclusion chromatography as a small peak (entries 2 and 3). The polymerization of 6 was carried out at 0 °C; however, poly 6 had a low Mn, which was indicative of the occurrence of some depolymerization, a typical reverse polymerization in a backbiting manner (entry 9). In contrast, the polymerization of monomer 7 with t-BuOK successfully proceeded even at 0 °C (entries 10 and 11). As mentioned above, the polymerization of 2 rapidly reached equilibrium even at low temperature. The low polymerizability of 3 and 4 was likely due to not only the poor solubilities of the monomer and polymer but also the steric hindrance of the substituent around the cyclic carbonate moiety.
Properties of polycarbonates
The polycarbonates (poly 2, poly 5, poly 6 and poly 7) showed relatively good solubility in typical organic solvents such as chloroform, toluene, DMF and dimethyl sulfoxide (Supplementary Table S1; see Supplementary Information) The good solubility can be understood in terms of the pendant fluorene moieties placed perpendicular to the main chain, which can inhibit the aggregation of polymer chains in a manner similar to 9,9-diarylfluorene-based cardo polymers.3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16 No diffraction peaks were observed by powder X-ray diffraction of the polymers, indicating their amorphous nature, which coincided with their observed good solubility (see Supplementary Information).
The thermal properties of the obtained polymers are summarized in Table 3. No melting points were confirmed, whereas the glass transition temperature (Tg) was observed. Poly 2 showed its Tg at 129 °C, which was lower than those of aromatic polycarbonates with alkyl spacers15, 19 but higher than the previously reported poly(2,2-disubstituted trimethylene carbonate)s.28, 55 Poly 6 had a Tg 185 °C, and the value was likely due to the rigid structure from the p-fluorophenyl group. The decomposition temperature (Td5) was approximately 300 °C for all of the polymers.
With the exception of poly 2, the film-forming abilities of the polymers were poor, likely because of their low molecular weights and higher contents of bulky aromatic rings. Then, the optical property of poly 2 in a film state was measured as a representative example. The thin film of poly 2 was prepared by casting the N-methylpyrrolidone solution onto a glass substrate followed by heating at 150 °C to dry the film (the solubility of poly 2 in ordinary low bp solvents was insufficient to prepare a clear cast film because of their high volatility. Therefore, we carried out the film preparation using high bp solvent such as NMP to which poly 2 has high solubility). The transmittance of poly 2 reached 90% at a wavelength of 380 nm and gradually increased with increasing wavelength over the visible light region. The high level of transparency observed for poly 2 resulted from its amorphous nature and was consistent with the significantly reduced levels of interchromophore interactions between or within the polymer chains. The refractive indices and birefringence values for poly 2 are summarized in Table 4. The refractive index at 587 nm was 1.620(6), which represented a high value for polymers consisting only of C, H and O atoms. The birefringence values of poly 2 were also evaluated in the undrawn state. The degree of retardation of visible light was found to be 0.0001 at 587 nm. The remarkably low level of birefringence could also be attributed to the vertical arrangement of the fluorene side groups relative to the main chains. The Abbe number of poly 2 was 23.45, which was low, as expected.
In summary, we have synthesized a series of novel fluorene-based cyclic carbonate monomers and obtained the corresponding cardo polymers by the anionic ROP of six-membered spirocyclic carbonate monomers. The polymers possessed fluorene skeletons placed perpendicular to the main chain. Interestingly 2 had a sufficient polymerizability to provide poly 2 with sufficiently high molecular weight in good yield, despite its structural similarity to the diphenyl-substituted cyclic carbonate with very low polymerizability. This result suggested that the connection of the two phenyl groups fairly enhanced the ROP of the six-membered cyclic carbonate. Volume expansion upon polymerization was confirmed with 2, which was in accordance with our previous report. In addition to 2, aromatic substituent-containing fluorene monomers were investigated their polymerizability. The 2,7-disubstituted fluorene monomers gave no polymers; however, the 3,6-disubstituted fluorene monomers afforded the corresponding polymers. All of the polymers obtained here exhibited good solubility in typical organic solvents, amorphous natures, favorable thermal stabilities, remarkably low birefringence values and high refractive indices. Thus, this study has demonstrated aliphatic polycarbonates having a fluorene moiety as cardo carbochain polymers with the characteristic optical property.

Synthesis and polymerization of 2.
References
Korshak, V. V., Vinogradova, S. V. & Vygodskii, Y. S. Cardo polymers. J. Macromol. Sci. Rev. Macromol. Chem. 11, 45–142 (1974).
Koyama, Y., Nakazono, K., Hayashi, H. & Takata, T. 9,9-Diarylfluorene moiety incorporated into polymer main chains: an essential skeleton exhibiting prominent physical, chemical, and optical properties. Chem. Lett. 39, 2–9 (2010).
Seesukphronrarak, S. & Takata, T. Novel fluorene-based biphenolic monomer: 9,9-bis(4-hydroxyphenyl)-9-silafluorene. Chem. Lett. 36, 1138–1139 (2007).
Seesukphronrarak, S. & Takata, T. Synthesis and characterization of novel poly(arylene thioether)s containing fluorene unit with high solubility and thermal stability. J. Polym. Sci. Pt. A: Polym. Chem. 45, 3073–3082 (2007).
Kawasaki, S., Yamada, M., Kobori, K., Jin, F., Kondo, Y., Hayashi, H., Suzuki, Y. & Takata, T. Synthesis and chemical, physical, and optical properties of 9,9-diarylfluorene-based poly(ether-ether-ketone). Macromolecules 40, 5284–5289 (2007).
Kawasaki, S., Yamada, M., Kobori, K., Yamada, M., Kakumoto, T., Tarutani, A. & Takata, T. Extraordinary carbon filler-dispersing power of fluorene-based polymers as matrix resins. Polym. J. 39, 115–117 (2007).
Seesukphronrarak, S., Kobori, K., Kawasaki, S. & Takata, T. Synthesis of properties of fluorene-containing poly(arylene sulfone)s. Polym. J. 39, 731–736 (2007).
Seesukphronrarak, S., Kawasaki, S. & Takata, T. Fluorene-rich high performance polyesters: synthesis and characterization of 9,9-fluorenylidene- and 2,7-fluorenylene-based polyesters with high refractive index and low birefringence. J. Polym. Sci. Pt. A: Polym. Chem. 46, 2549–2556 (2008).
Kawasaki, S., Yamada, M., Kobori, K., Jin, F. & Takata, T. Fine dispersion of carbon black in fluorene-based resin. Polym. Composit. 29, 1044–1048 (2008).
Kawasaki, S., Yamada, M., Kobori, K., Sakamoto, H., Kondo, Y., Jin, F. & Takata, T. Preparation of a novel alloy composed of fluorene-based polyester and polycarbonate and their properties for the optical use. J. Appl. Polym. Sci. 111, 461–468 (2009).
Hayashi, H., Kawasaki, S., Kobori, K., Koyama, Y., Asai, S. & Takata, T. Synthesis and properties of polysiloxanes possessing 9,9-diarylfluorene structure in the main chain. Polym. J. 41, 272–278 (2009).
Hayashi, H. & Takata, T. 9,9-Diarylfluorene-based poly(alkyl aryl ether)s: synthesis and property. Polym. J. 41, 609–615 (2009).
Kawasaki, S., Kato, M., Kobori, K., Sakamoto, H., Kondo, Y. & Takata, T. New concept of reducing a birefringence of poly(ethylene naphthalate) by a novel alloy with fluorene-based polyester. Polym. Eng. Sci. 49, 2374–2383 (2009).
Kawasaki, S., Yamada, M., Kobori, K., Jin, F. & Takata, T. High dispersion ability of fluorene-based polyester as matrix polymer for carbon nanotubes and carbon fibers. J. Appl. Polym. Sci. 118, 2690–2695 (2010).
Hasegawa, T., Koyama, Y., Seto, R., Kojima, T., Hosokawa, K. & Takata, T. Diphenolic 9,9-diarylfluorene trimers and derivatives possessing flexible alkylene chain spacers: synthesis of the monomers, their polymerization, and properties of the resulting polymers. Macromolecules 43, 131–136 (2010).
Okuda, H., Koyama, Y. & Takata, T. 9,9-Diaryl-4,5-diazafluorene-based cardo polymer; synthesis and characteristic properties. J. Polym. Sci. Pt. A: Polym. Chem. 51, 4541–4549 (2013).
Seto, R., Kojima, T., Hosokawa, K., Koyama, Y., Konishi, G. & Takata, T. Synthesis and property of 9,9'-spirobifluorene-containing aromatic polyesters as optical polymers with high refractive index and low birefringence. Polymer 51, 4744–4749 (2010).
Okuda, H., Seto, R., Koyama, Y. & Takata, T. Poly(arylene thioether)s containing 9,9'-spirobifluorene moieties in the main chain: masked dithiol-based synthesis and excellent optical properties. J. Polym. Sci. Pt. A: Polym. Chem. 48, 4192–4199 (2010).
Seto, R., Sato, T., Kojima, T., Hosokawa, K., Koyama, Y., Konishi, G. & Takata, T. 9,9'-Spirobifluorene-containing polycarbonates: transparent polymers with high refractive index and low birefringence. J. Polym. Sci. Pt. A: Polym. Chem. 48, 3658–3667 (2010).
Okuda, H., Seto, R., Koyama, Y. & Takata, T. Poly(arylene sulfone)s containing 9,9'-spirobifluorene: synthesis and excellent optical properties. Polym. J. 42, 795–798 (2010).
Ishiwari, F., Kawasaki, S., Koyama, Y. & Takata, T. Poly(vinylspirobifluorene): synthesis and property of a novel styrenic polymer. Chem. Lett. 37, 853–854 (2008).
Mei, J., Aitken, B. S., Graham, K. R., Wagener, K. B. & Reynolds, J. R. Regioregular electroactive polyolefins with precisely sequenced π-conjugated chromophores. Macromolecules 43, 5909–5913 (2010).
Alder, R. W., Anderson, K. R., Benjes, P. A., Butts, C. P., Koutentis, P. A. & Orpen, A. G. Polymers and oligomers with transverse aromatic groups and tightly controlled chain conformations. Chem. Commun. 24, 309–310 (1998).
Braun, A. M., Cassidy, H. G., Schulz, R. C. & Tanaka, H. Charge-transfer complexes of polymers from substituted spirocyclopropane-1,9'-fluorene compounds. 1. Model compounds and oligomers. Makromol. Chem. 146, 195–205 (1971).
Nakano, T. Synthesis, structure and function of π-stacked polymers. Polym. J. 42, 103–123 (2010).
Wang, Y., Sakamoto, T. & Nakano, T. Molecular chirality induction to an achiral n-conjugated polymer by circularly polarized light. Chem. Commun. 48, 1871–1873 (2012).
Watanabe, K., Sakamoto, T., Taguchi, M., Fujiki, M. & Nakano, T. A chiral π-stacked vinyl polymer emitting white circularly polarized light. Chem. Commun. 47, 10996–10998 (2011).
Endo, T., Shibasaki, Y. & Sanda, F. Controlled ring-opening polymerization of cyclic carbonates and lactones by an activated monomer mechanism. J. Polym. Sci. Pt. A: Polym. Chem 40, 2190–2198 (2002).
Sekiguchi H . Ring-Opening Polymerization (eds Ivan K. J., Saegusa T). vol. 2, (Elsevier, London, UK, 1984).
Kamber, N. E., Jeong, W., Waymouth, R. M., Pratt, R. C., Lohmeijer, B. G. G. & Hedrick, J. L. Organocatalytic ring-opening polymerization. Chem. Rev. 107, 5813–5840 (2007).
Matsuo, J., Aoki, K., Sanda, F. & Endo, T. Substituent effect on the anionic equilibrium polymerization of six-membered cyclic carbonates. Macromolecules 31, 4432–4438 (1998).
Matsuo, J., Sanda, F. & Endo, T. A novel observation in anionic ring-opening polymerization behavior of cyclic carbonates having aromatic substituents. Macromol. Chem. Phys. 199, 2489–2494 (1998).
Takata, T. & Endo, T. Recent advances in the development of expanding monomers: synthesis, polymerization and volume change. Prog. Polym. Sci. 18, 839–870 (1993).
Takata., T., Sanda, F., Ariga, T., Nemoto, H. & Endo, T. Cyclic carbonates, novel expandable monomers on polymerization. Macromol. Rapid Commun. 18, 461–469 (1997).
Takata, T. & Endo, T. in Expanding Monomers-Synthesis, Characterization, and Application (eds Sadhir R. K., Luck R. M). Ch. 3, 63–152 (CRC Press, Florida, USA, 1992).
Ariga, T., Takata, T. & Endo, T. Cationic ring-opening polymerization of cyclic carbonates with alkyl halides to yield polycarbonate without the ether unit by suppression of elimination of carbon dioxide. Macromolecules 30, 737–744 (1997).
Kakimoto, K., Nemoto, N., Sanda, F. & Endo, T. Anionic ring-opening polymerization of cyclic thiocarbonates containing norbornene and norbornane groups undergoing volume expansion on polymerization. Chem. Lett. 2, 156–157 (2002).
Kricheldorf, H. R., Dunsing, R. & Serra, A. Polylactones. 12. Cationic polymerization of 2,2-dimethyltrimethylene carbonate. Makromol. Chem. 188, 2453–2466 (1987).
Albertsson, A.-C. & Sjoeling, M. Homopolymerization of 1,3-dioxan-2-one to high-molecular-weight poly(trimethylene carbonate). J. Macromol. Sci., Pure Appl. Chem. A29, 43–54 (1992).
Keul, H., Bächer, R. & Höcker, H. Anionic ring-opening polymerization of 2,2-dimethyltrimethylene carbonate. Makromol. Chem. 187, 2579–2589 (1986).
Kricheldorf, H. R., Jenssen, J. & Kreiser-Sanders, I. Polymers of carbonic acid. 6. Polymerization of trimethylene carbonate (1,3-dioxan-2-one) with complexation catalysts. Makromol. Chem. 192, 2391–2399 (1991).
Carter, K. R., Richter, R., Kricheldorf, H. R. & Hedrick, J. L. Synthesis of amine-terminated aliphatic polycarbonates via Al(Et)2(OR)-initiated polymerizations. Macromolecules 30, 6074–6076 (1997).
Dobrzynski, P. & Kasperczyk, J. Synthesis of biodegradable copolymers with low-toxicity zirconium compounds. V. Multiblock and random copolymers of L-lactide with trimethylene carbonate obtained in copolymerizations initiated with zirconium(IV) acetylacetonate. J. Polym. Sci. Pt. A: Polym. Chem. 44, 3184–3201 (2006).
Bisht, K. S., Svirkin, Y. Y., Henderson, L. A., Gross, R. A., Kaplan, D. L. & Swift, G. Lipase-catalyzed ring-opening polymerization of trimethylene carbonate. Macromolecules 30, 7735–7742 (1997).
Kricheldorf, H. R., Lee, S.-R. & Weegen-Schulz, B. Polymers of carbonic acid. 12. Spontaneous and hematin-initiated polymerizations of trimethylene carbonate and neopentylene carbonate. Macromol. Chem. Phys. 197, 1043–1054 (1996).
Varma, I. K., Albertsson, A.-C., Rajkhowa, R. & Srivastava, R. K. Enzyme catalyzed synthesis of polyesters. Prog. Polym. Sci. 30, 949–981 (2005).
Yamashita, C., Nakazono, K. & Takata, T. Ring-opening polymerization of fluorene skeleton-containing spirocyclic monomer and property of the resulting polymers. Polym. Prepr., Jpn 60, 512 (2011).
Venkataraman, S., Hedrick, J. L. & Yang, Y. Fluorene-functionalized aliphatic polycarbonates: design, synthesis and aqueous self-assembly of amphiphilic block copolymers. Polym. Chem. 5, 2035–2040 (2014).
Eugene, G. Reactions of active methylene compounds in pyridine solution II. Aldol-type reactions of indene and fluorene. J. Am. Chem. Soc. 82, 4945–4952 (1960).
Young, R. N., Quirk, R. P. & Fetters, L. J. Anionic Polymerizations of Nonpolar Monomers Involving Lithium (eds Cantow H.-J., Fetters L. J). 3–90, (Springer-Verlag, New York, NY, USA, 1984).
Brown, T. L., Ladd, J. A. & Newman, G. N. Interaction of alkyllithium compounds with base: complex formation between ethyllithium and lithium ethoxide in hydrocarbon solvents. J. Organomet. Chem. 3, 1 (1965).
Bywater, S. & Worsfold, D. J. Alkyllithium anionic polymerization initiators in hydrocarbon solvents. J. Organomet. Chem. 10, 1–6 (1967).
Hsieh, H. L. Kinetics of polymerization of butadiene, isoprene, and styrene with alkyllithiums I. Rate of polymerization. J. Polym. Sci., A-3, 153–161 (1965).
Hsieh, H. L. Kinetics of polymerization of butadiene, isoprene, and styrene with alkyllithiums II. Rate of initiation. J. Polym. Sci. A-3, 163–172 (1965).
Rokicki, G. Aliphatic cyclic carbonates and spiroorthocarbonates as monomers. Prog. Polym. Sci. 25, 259–342 (2000).
Acknowledgements
This work was financially supported in part by the Tokuyama Science Foundation and the Global COE Program (Education and Research Center for Material Innovation), MEXT, Japan. We give special thanks to Dr Takahiro Kojima and Mr Katsumoto Hosokawa from the Production Engineering Research Laboratory, Canon Inc., for their help in the measurement of the optical properties.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Polymer Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Nakazono, K., Yamashita, C., Ogawa, T. et al. Synthesis and properties of pendant fluorene moiety-tethered aliphatic polycarbonates. Polym J 47, 355–361 (2015). https://doi.org/10.1038/pj.2015.7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pj.2015.7
