
Abstract
Human sterile α motif and HD domain-containing protein 1 (SAMHD1) has deoxyribonucleoside triphosphohydrolase (dNTPase) activity that allows it to defend against human immunodeficiency virus type I (HIV-1) infections and regulate the cell cycle. Although SAMHD1 mutations have been identified in various cancer types, their role in cancer is unclear. Here, we aimed to investigate the oncogenic role of SAMHD1 in human clear cell renal cell carcinoma (ccRCC), particularly as a core molecule promoting cancer cell migration. We found that SAMHD1 participated in endocytosis and lamellipodia formation. Mechanistically, SAMHD1 contributed to the formation of the endosomal complex by binding to cortactin. Thereafter, SAMHD1-stimulated endosomal focal adhesion kinase (FAK) signaling activated Rac1, which promoted lamellipodia formation on the plasma membrane and enhanced the motility of ccRCC cells. Finally, we observed a strong correlation between SAMHD1 expression and the activation of FAK and cortactin in tumor tissues obtained from patients with ccRCC. In brief, these findings reveal that SAMHD1 is an oncogene that plays a pivotal role in ccRCC cell migration through the endosomal FAK-Rac1 signaling pathway.
Similar content being viewed by others


Introduction
Human sterile α motif and HD domain-containing protein 1 (SAMHD1) was originally identified as a protein whose mutation is related to Aicardi–Goutières syndrome, which is an autoimmune disease1. Later, SAMHD1 was discovered to act as a deoxyribonucleoside triphosphohydrolase (dNTPase) in mammals. The most well-studied biological functions of SAMHD1 are involved in the early immune response against viruses, such as human immunodeficiency virus type I (HIV-I), hepatitis B virus, and herpes simplex virus 1. Previous studies revealed that SAMHD1 hydrolyzes intracellular dNTPs—essential components for viral reverse transcription or DNA replication—at the early stage of viral infection1,2,3,4. Since several previous studies have observed SAMHD1 nuclear accumulation1,5,6, SAMHD1 was presumed to function in the nucleus. However, inactivation of the nuclear localization sequence (NLS) affects neither the dNTPase nor the restriction activities of SAMHD1, demonstrating that nuclear localization is not a prerequisite for these functions7,8. Cytoplasmic SAMHD1 is protected from Vpx-mediated proteasomal degradation and can prevent HIV-I infection5,8. A previous study reported that SAMHD1 is translocated from the nucleus to the cytoplasm through growth factor stimulation9, suggesting that SAMHD1 may perform different functions depending on its intracellular location.
Recently, SAMHD1 was reported to be associated with cancer progression. In total, 1 542 SAMHD1 mutations have been identified in various cancer types, including chronic lymphocytic leukemia, mantle cell lymphoma, and colorectal cancer10,11,12. SAMHD1 promotes chemoresistance because, given the structural similarity to endogenous dNTP, the nucleotide analogs used for acute myeloid leukemia and mantle cell lymphoma treatments are degraded by SAMHD113,14,15. In contrast, cell proliferation in lung adenocarcinoma (LUAD) is suppressed by SAMHD1 overexpression16. Therefore, the roles of SAMHD1 in cancer are unclear and controversial. Furthermore, the role of SAMHD1 in tumorigenesis and the underlying mechanisms of SAMHD1-mediated cancer progression remain unknown.
During cell migration, the plasma membrane functions as a central site for adhesion formation, force generation, and signal transduction17,18,19. The main factors determining cell migration are membrane protrusion and focal adhesion or membrane–matrix adhesion20. In focal adhesion, focal adhesion kinase (FAK) is activated through autophosphorylation and forms an active FAK-Src complex21. The FAK-Src complex phosphorylates proteins associated with nascent adhesion and finally recruits active Rac1 to the membrane ruffles and adhesion sites22. Activated Rac1 accumulates cortactin at the cell periphery to form lamellipodia with wide sheet-like membrane structures. Cortactin binds to actin filaments formed on the lamellipodia to stabilize polymerized actin23,24. In the late adhesion stage, Rac1 activity is diminished, whereas Rho A—a member of the Rho GTPase family proteins—is activated to promote propulsion and mature focal adhesion25,26. Finally, cortactin phosphorylation aids the depolymerization of actin filaments on the lamellipodia so that the focal adhesions of the protruding membrane can be separated to promote cell motility27,28.
Sufficient biomass supplementation is important to sustain focal adhesion of the plasma membrane during cell migration. Cancer cells have adapted an endosomal trafficking system to effectively supply materials during metastasis. Endocytosis supplies important plasma membrane components of moving cells that maintain cell polarity and facilitate cell migration29,30. Rather than being degraded within the cell, membrane receptors are rapidly recycled from the cell membrane through endo- and exocytosis. Endocytosis proceeds with the stepwise complex formation of several proteins, including FAK and cortactin. Subsequently, Rab GTPases and early endosome antigen 1 (EEA1) wrap around the endosome–protein complex to mark the target vesicle31,32. The expression of this recycling system is reported to be positively correlated with cell motility and cancer metastasis31. Increasing evidence indicates that cell migration and endocytosis occur concomitantly; however, the precise mechanism of endocytosis activation and endosomal membrane assembly remains elusive.
This study aimed to elucidate the mechanism by which SAMHD1 regulates cell migration in clear cell renal cell carcinoma (ccRCC). We identified SAMHD1 as a new binding partner of cortactin in early endosomes and lamellipodia. Moreover, SAMHD1-mediated endosomal FAK signaling stimulated Rac1 activation to trigger lamellipodial protrusion, promoting cell motility. Collectively, our study indicated that SAMHD1 modulates ccRCC cell migration through endosomal regulation and identified SAMHD1 as a potential therapeutic target for ccRCC treatment.
Materials and methods
Cell culture and reagents
HK-2 (#22190, a normal kidney cell line), Caki-1 (#30046), Caki-2 (#30047), SN12C (#80025), and ACHN (#21611) cells were obtained from the Korean Cell Line Bank (Seoul, Korea). Two RCC cell lines with lower SAMHD1 expression were used for SAMHD1 overexpression experiments, and two other RCC cell lines with higher SAMHD1 expression were used for SAMHD1 knockdown experiments. HK-2 cells were grown in RPMI 1640, and the other cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin in a humidified atmosphere of 5% CO2 at 37 °C. PF573228 (#PZ0117) and fasudil (#CDS021620) were purchased from Sigma‒Aldrich (St. Louis, MO, USA). NSC23766 (#S8301), ML141 (#S7686), and Dynasore (#S8047) were obtained from Selleckchem (Houston, TX, USA).
Transfection and stable cell line construction
SN12C and ACHN cells were transfected with siRNA targeting SAMHD1 (5′- GAUUCAUUGUGGCCAUAUA-3′) using Oligofectamine Transfection reagent (#12252011, Invitrogen, Gaithersburg, MD, USA), and Caki-2 cells were transfected with pCDNA3.1-FLAG-SAMHD1 using Lipofectamine 2000 (#11668019, Invitrogen) according to the manufacturer’s instructions.
To establish stable Caki-1 cells, pIRES-FLAG-SAMHD1 plasmids were transfected into HEK293T cells with the lentivirus packing vectors psPAX2 (Addgene, Cambridge, MA, USA) and pMD2. G (Addgene), and retroviral supernatants were harvested after 48 h. Caki-1 cells were transduced with the collected supernatants, and the transduced cells were selected for one month using blasticidin (10 μg·ml–1, Sigma‒Aldrich). The expression of FLAG-SAMHD1 was quantified using western blotting.
Antibodies
An anti-SAMHD1 antibody (OTI1A1, ab128107, 1:2 000) and an anti-phosphorylated cortactin Y421 antibody (ab47768, 1:1 000) were purchased from Abcam (Cambridge, UK). Anti-E-cadherin (24E10, #3195, 1:1 000), anti-N-cadherin (13A9, #14215, 1:1 000), anti-vimentin (R28, #3932, 1:2 000), anti-snail1 (C15D3, #3879, 1:1 000), anti-ZEB1 (D80D3, #3396, 1:1 000), anti-FAK (D5O7U, #71433, 1:2 000), anti-phosphorylated FAK (Y397; D20B1, #8556, 1:1 000), and anti-EEA1 antibodies (C45B10, #3288, 1:1 000) were purchased from Cell Signaling Technology (Beverly, MA, USA). Anti-cortactin (4F11, 05-180, 1:2 000) and anti-paxillin antibodies (5H11, 05-417, 1:200) were purchased from MilliporeSigma (Burlington, MA, USA). Anti-fibronectin (F3648, 1:1 000), anti-FLAG (M2, F1804, 1:2 000), anti-β-actin (A2066, 1:2 000), and anti-α-tubulin antibodies (DM1A, T9026, 1:2 000) were purchased from Sigma‒Aldrich. For immunohistochemistry, an anti-SAMHD1 antibody (NBP1-31432, 1:50) was purchased from Novus Biologicals (Centennial, CO, USA). An anti-phosphorylated FAK (Y397) antibody (#700255, 1:50) was purchased from Invitrogen (Gaithersburg, MD, USA), and an anti-phosphorylated cortactin (Y421) antibody (LS‑C353975, 1:100) was purchased from LSBio (Seattle, WA, USA).
The Cancer Genome Atlas (TCGA) transcriptome data analysis
From the Broad GDAC Firehose database (https://gdac.broadinstitute.org/), we downloaded RNA-sequencing (RNA-seq) and clinical data from the pankidney cohort (KIPAN), clear cell renal cell carcinoma (KIRC, ccRCC in the present study), renal papillary cell carcinoma (KIRP, PRCC in the present study), and kidney chromophobe (KICH, chRCC in the present study) datasets. In addition, we downloaded the GBMLGG, UCEC, LIHC, THCA, COADREAD, BLCA, LUAD, and LUSC datasets from TCGA. mRNA expression data were used to analyze SAMHD1 expression and epithelial–mesenchymal transition (EMT)-related genes. Gene-set enrichment analysis (GSEA) was performed using GSEA software provided by the University of California San Diego and Broad Institute33. Gene Ontology (GO) analysis was performed using the same software. We only considered results with a p value < 0.05 significant.
Survival analysis for RCC subtypes
The RNA-Seq whole genome expression profile, corresponding clinical parameters, and follow-up information for KIRC, KIRP, and KICH were downloaded as FPKM-UQ-normalized gene expression data from the UCSC XENA database (https://xenabrowser.net/). For the analysis of SAMHD1, each subtype of RCC was divided into “SAMHD1-high” (top 20%) and “SAMHD1-low” (bottom 80%) mRNA expression groups. For the analysis of the 5-year survival rate, survival data for the high and low expression groups were retrieved from the UCSC Xena database and analyzed using GraphPad Prism V8.2.0 (GraphPad Software, San Diego, CA, USA). Kaplan–Meier plots were used to analyze the 5-year overall survival (OS) and progression-free survival (PFS) rates of RCC patients in TCGA. Log-rank tests for significance and Kaplan–Meier curves were generated using GraphPad Prism V8.2.0 with statistical significance set at p < 0.05. A univariate Cox regression analysis was performed to examine the relationship between SAMHD1 expression and OS to determine the functional significance of SAMHD1 in RCC prognosis.
Immunoblotting and immunoprecipitation
Proteins were extracted using a lysis buffer from Cell Signaling Technology (#9803) with a protease inhibitor cocktail (Gendepot, Katy, TX, USA), and 30 μg of the lysates were used for immunoblotting. For immunoprecipitation, 900 μg protein was incubated overnight at 4 °C with the corresponding primary antibodies conjugated to G beads (MilliporeSigma) or FLAG-tagged magnetic beads (Sigma‒Aldrich). Beads were washed with a washing buffer containing 20 mM Tris (pH 8.0), 150 mM NaCl, 0.2 mM EDTA, and 0.1% Triton X-100. After SDS‒PAGE, membranes were immunoblotted using the corresponding primary antibodies and HRP-conjugated secondary antibodies. Visualization was performed using Lumi-Pico solution (Dogen, Seoul, Korea) and a LAS-4000 instrument (GE Healthcare, Chicago, IL, USA). SAMHD1 levels were quantified as integrated band intensities using ImageJ (National Institutes of Health, Bethesda, MD, USA) and normalized to the levels of the corresponding housekeeping proteins.
Cell migration assay
Cells were plated in a 6-well plate and cultured until 100% confluency was reached. Wounds were made using a 200 μL pipette tip and washed with PBS. To reduce the influence on cell proliferation, cells were treated with 5 μM thymidine. After 24 h of incubation, the wound closure rate was measured using a phase-contrast microscope.
Transwell migration and invasion assay
Transwell chambers (#3422, Corning, Summerville, MA, USA) were used with or without Matrigel (#354234, Corning) to perform cell migration or invasion assays. ccRCC cells (1–2.5 × 104) were resuspended in serum-free medium and plated onto the inner chambers. DMEM with 10% FBS was added to the outer chambers. The cells were set aside for 24 h to allow migration and invasion before the inserts were removed and washed with PBS. Cells that migrated to or invaded the opposite side of the inserts were stained with 0.5% crystal violet; the number of cells was counted using ImageJ.
RNA isolation and mRNA analysis
Total RNA was extracted using TRIzol reagent (Invitrogen) and reverse-transcribed into cDNA using Moloney murine leukemia virus (MMLV) reverse transcriptase (Promega, Madison, WI, USA). Quantitative real-time PCR was performed using a LightCycler 480 Instrument II (Roche, Basel, Switzerland). The comparative Ct (2-ΔΔCt) method was used to quantify the mRNA levels, which were normalized to the levels of the corresponding housekeeping genes.
Immunocytochemistry
Immunocytochemistry was performed as previously described34. Briefly, ccRCC cells were seeded onto glass coverslips in 24-well plates (4 × 104 cells per well). Cells were fixed in 4% paraformaldehyde for 15 min. After washing with PBS, cells were permeabilized in PBS containing 0.1% Triton X-100 for 10 min, blocked with 3% normal goat serum for 1 h, and then incubated with primary antibodies for 1 h. After washing with PBS, the coverslips were incubated with Alexa-488 or 546-conjugated secondary antibodies (Invitrogen) for 1 h at room temperature followed by counterstaining with 4′,6-diamidino-2-phenylindole (Sigma‒Aldrich). ProLong Gold Antifade Mountant (Invitrogen) was used for mounting, fluorescence images were obtained using confocal microscopy (LSM5 EXCITER, Carl Zeiss, AG, Germany), and immunopositive cellular structures were quantified using ImageJ.
Immunohistochemistry
Frozen human ccRCC tissues were cut into 10 μm sections using a cryostat microtome (CM1860, Leica, Wetzlar, Germany). The sections were fixed in 100% acetone for 20 min at −20 °C and blocked with 3% goat serum for 1 h. The sections were incubated with primary antibodies overnight at 4 °C. After washing with PBS, the sections were incubated with biotin-labeled secondary antibodies (Vector Labs, Burlingame, CA, USA) and staining reactions were carried out using a VECTASTAIN Elite ABC HRP Kit (PK-6101, Vector Labs) according to the manufacturer’s instructions. The immunoreaction sections were visualized with ImmPACT DAB Substrate Peroxidase (SK-4105, Vector Labs) and counterstained with hematoxylin. Quantitative analysis of the SAMHD1-, p-Cortactin-, and p-FAK-positive areas (with brown staining) was performed with ImageJ (NIH) using the protocol provided by the LSU Health Sciences Center35. Images of the stained tissues were adjusted relative to the threshold using the RGB mode.
ccRCC clinical specimen collection
We analyzed 20 pairs of human tissues (tumor and normal specimens) from Dongsan Hospital, Daegu, Korea. All specimens were obtained from patients who provided informed consent as stipulated by the Institutional Review Board at Keimyung University (School of Medicine), and the study protocol was approved by the ethics committee of Keimyung University Hospital (#2020-10-068). The tissues were subjected to western blotting and immunohistochemistry to compare SAMHD1, phospho-FAK, and phospho-cortactin levels between the matched samples.
Statistical analysis
The results are expressed as the mean ± SD or SEM. We used a two-tailed Student’s t test for single comparisons or two-way ANOVA for multiple comparisons. Statistical significance was set at p < 0.05. GraphPad Prism V8.2.0 (GraphPad Software) was used for statistical analyses.
Results
High SAMHD1 expression is associated with poor prognosis in ccRCC
To elucidate the role of SAMHD1 in cancer, TCGA datasets were used to analyze the expression of SAMHD1 in different cancer types. Among the several types of cancer, the difference in SAMHD1 expression between cancerous and normal tissues was the highest in pankidney cancer (Fig. 1a). RCC is classified into three subtypes according to histological characteristics36. Accordingly, we compared SAMHD1 mRNA levels between three RCC subtype datasets. Interestingly, only ccRCC had significantly upregulated mean SAMHD1 mRNA expression in tumor tissues compared with normal tissues (Fig. 1b). SAMHD1 mRNA levels were higher in tumor tissues than in adjacent normal tissues from patients with the ccRCC subtype (Fig. 1c). Furthermore, we investigated the relationship between prognosis and SAMHD1 expression in patients with ccRCC. Notably, Kaplan–Meier survival analysis revealed that patients with high SAMHD1 expression exhibited worse OS and PFS rates (Fig. 1d, e) than patients with low SAMHD1 levels. We did not observe any relationship between SAMHD1 expression and OS or PFS in PRCC and chRCC (Supplementary Fig. 1). Immunohistochemistry and western blot analysis showed higher protein expression levels of SAMHD1 in tumor tissues than in the paired normal tissues (Fig. 1f, g). Analysis of protein levels according to cancer grades indicated that SAMHD1 was overexpressed in tumor tissues with intermediate and poor prognoses. The protein level of SAMHD1 was also increased in tumor tissues with favorable prognoses compared to their adjacent normal tissues, but there was no statistically significant difference (Fig. 1h, i). Since the oncogenic effects of SAMHD1 mutations have already been reported in some cancer types, we investigated the occurrence of SAMHD1 mutations in RCC using the TCGA database. SAMHD1 mutations rarely occurred in RCC; only 4 out of 330 patients with ccRCC and 1 out of 277 patients with PRCC exhibited SAMHD1 mutations. Among the mutations, two were deletions, and three were substitutions (data not shown). Collectively, these results suggest that SAMHD1 expression may be associated with ccRCC development.

a Comparing the gene expression of SAMHD1 in various cancer types using the TCGA database. KIPAN, pankidney cancer; GBMLGG, glioma; UCEC, uterine corpus endometrial carcinoma; LIHC, liver hepatocellular carcinoma; THCA, thyroid carcinoma; COADREAD, colorectal adenocarcinoma; BLCA, bladder urothelial carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma. b Comparison of SAMHD1 mRNA expression between tumor and normal tissues according to histological RCC subtypes. ***p < 0.001. c Comparative mRNA expression of SAMHD1 between paired tumor and nontumor tissues according to histological RCC subtypes. ***p < 0.001. d Kaplan–Meier analysis of 5-year overall survival (OS) between SAMHD1 high expression (n = 77) and SAMHD1 low expression groups (n = 303). Log-rank p = 0.004. e Kaplan–Meier analysis of progression-free survival (PFS) between SAMHD1 high expression (n = 78) and SAMHD1 low expression groups (n = 300). Log-rank p = 0.002. f Immunohistochemistry (IHC) of SAMHD1 in ccRCC tissues. Scale bar, 50 μm. g Immunoblotting was used to analyze the protein level of SAMHD1 in 20 paired cancer and normal tissues. SAMHD1 protein levels were normalized to corresponding β-actin levels. **p < 0.01. h SAMHD1 protein levels normalized to β-actin levels in ccRCC tissues according to patient prognosis were analyzed by western blotting. *p < 0.05. Data are presented as the mean ± SD. i Representative western blot bands are presented with the corresponding prognosis.
SAMHD1 potentiates the migration and invasion of ccRCC cells
To study the role of SAMHD1 in ccRCC, we first assessed the basal level of SAMHD1 expression in four ccRCC cell lines compared with that in the normal kidney epithelial cell line HK-2 (Fig. 2a). Based on the basal level of SAMHD1 in each cell line, Caki-1 and Caki-2 cells were used for SAMHD1 overexpression, and SN12C and ACHN cells were used for knockdown experiments. Analysis of cell proliferation ability indicated that SAMHD1 expression did not influence proliferation (Supplementary Fig. 2a, b). Subsequently, we examined the effect of SAMHD1 on cell migration using in vitro wound-healing assays and found that SAMHD1 knockdown suppressed cell migration in both the ACHN and SN12C cell lines (Fig. 2b), whereas SAMHD1 overexpression significantly promoted the migration of ccRCC cells (Fig. 2c). The transwell migration assay also showed consistent results (Fig. 2d, e). The invasion assay confirmed that SAMHD1 knockdown markedly inhibited the invasiveness of ACHN and SN12C cells (Fig. 2f), whereas exogenous SAMHD1 overexpression facilitated the invasiveness of Caki-1 and Caki-2 cells compared with control cells (Fig. 2g). These results indicate that SAMHD1 facilitates ccRCC cell migration.

a Western blot analysis of SAMHD1 expression in normal renal epithelial cells (HK-2) and different RCC cell lines (Caki-1, Caki-2, SN12C, ACHN). Densitometric analysis of SAMHD1 protein expression levels relative to β-actin levels. b, c Wound-healing analysis of cell migration ability in SAMHD1-knockdown ACHN and SN12C cells or SAMHD1-overexpressed Caki-1 and Caki-2 cells over 24 h. Data are presented as the mean ± SD. The data shown are representative of three independent experiments. **p < 0.01; ***p < 0.001. d, e Transwell migration assay of cell migration capacity upon SAMHD1 knockdown in ACHN and SN12C cells or overexpression in Caki-1 and Caki-2 cells over 24 h. Data are presented as the mean ± SD. The data shown are representative of three independent experiments. ***p < 0.001. f, g Analysis of cell invasion ability upon SAMHD1 knockdown in ACHN and SN12C cells or overexpression in Caki-1 and Caki-2 cells. *p < 0.05; ***p < 0.001. Data are presented as the mean ± SD of three independent experiments.
SAMHD1 enhances ccRCC cell mesenchymal characteristics to increase migration capacity
Interestingly, we noted SAMHD1-induced changes in cell morphology that promoted cell migration or invasion depending on the SAMHD1 level. ACHN and SN12C cells could not form tips at their edges of SAMHD1 knockdown cells (Fig. 3a); conversely, when SAMHD1 was overexpressed in Caki-1 and Caki-2 cells, the cell body became pointed and appeared to be directional. Based on these findings, we hypothesized that SAMHD1 regulates cell migration through epithelial–mesenchymal transition, a well-known mechanism by which cancer cells acquire mesenchymal characteristics and increase motility by losing cell–cell junctions and basal polarity37. SAMHD1 knockdown upregulated the expression of E-cadherin—an epithelial marker—and downregulated that of mesenchymal markers, such as fibronectin, vimentin, snail1, and ZEB1 (Fig. 3b, c). In contrast, the opposite effects were observed following SAMHD1 overexpression (Fig. 3d, e). Furthermore, mRNA analysis of TCGA datasets showed consistent results with those obtained from western blotting and quantitative real-time PCR experiments (Fig. 3f). Similar to the in vitro results, E-cadherin expression was downregulated in tumor tissues compared to normal tissues of patients with ccRCC (Fig. 3g–i). Overall, these results suggest that SAMHD1 promotes EMT and enhances the mesenchymal characteristics required for effective ccRCC cell migration.

a Phase-contrast microscopy revealed changes in cell morphology from epithelial to mesenchymal characteristics in different ccRCC cells after SAMHD1 knockdown or overexpression. Scale bar, 20 μm. b Western blot analysis of epithelial and mesenchymal gene expression after SAMHD1 knockdown in ACHN and SN12C cells. c qPCR analysis of mesenchymal markers after SAMHD1 knockdown in SN12C cells. Data are presented as the mean ± SD. The experiments were independently performed at least three times. ***p < 0.001. d Western blot analysis of epithelial and mesenchymal markers in SAMHD1-overexpressing Caki-1 and Caki-2 cells. e qPCR analysis of mesenchymal markers in SAMHD1-overexpressed Caki-1 cells. Data are presented as the mean ± SD. The data shown are representative of three independent experiments. ***p < 0.001. f Association between the expression of SAMHD1 and that of epithelial or mesenchymal markers based on ccRCC clinical data. g E-cadherin protein levels relative to β-actin levels for 20 paired tumor and normal tissues. Data are presented as the mean ± SD. ***p < 0.001. h E-cadherin protein levels relative to β-actin levels in tissues of patients with ccRCC according to prognosis. Data are presented as the mean ± SD. **p < 0.01; ***p < 0.001. i Western blot analysis of E-cadherin expression in 20 paired tissue samples from patients with ccRCC. Western blot bands are presented with the corresponding prognosis. Data are presented as the mean ± SD.
SAMHD1 promotes the formation of lamellipodia in ccRCC cells
Given that SAMHD1 acts as an oncogene and accelerates ccRCC cell migration, we hypothesized that SAMHD1 could positively regulate cell migration. Therefore, GSEA was performed using the ccRCC dataset from TCGA (dataset was downloaded through https://gdac.broadinstitute.org) to elucidate the detailed mechanism by which SAMHD1 regulates cell migration. The enriched GO subcategories indicated that the genes upregulated upon SAMHD1 overexpression were mainly associated with the structural features of actively migrating cells (Fig. 4a). Furthermore, we found that lamellipodia and membrane ruffles—subcategories in the cellular component category—were highly enriched (Fig. 4b, c). To further validate the GSEA results, we performed an immunofluorescence analysis of paxillin, which participates in nascent cell adhesion, spreading, and migration through interactions with multiple structural proteins38. Many paxillin-positive signals were observed at the cell periphery in the presence of SAMHD1 (Fig. 4d); however, paxillin-positive signals were barely present in SAMHD1-knockdown SN12C cells. Consistently, more paxillin-positive signals were observed in SAMHD1-overexpressing Caki-1 cells than in control cells (Fig. 4e). To assess the impact of SAMHD1 on lamellipodium or membrane ruffle development, ccRCC cells were transfected with RFP-cortactin, a well-known lamellipodium marker. As shown in Fig. 4f, the lamellipodium area was significantly reduced in SAMHD1-deficient cells compared with control cells. In contrast, SAMHD1-overexpressing Caki-1 cells had a larger lamellipodia area and stronger cortactin signals than those in control cells (Fig. 4g). Our findings suggest that SAMHD1 promotes lamellipodia formation during the nascent stage of cell migration.

a Gene Ontology (GO) analysis of the cellular component subcategories positively regulated by SAMHD1 overexpression. b, c Gene set enrichment analysis (GSEA) related to cytoskeletal changes for cell migration in the SAMHD1 high expression group. ***p < 0.001. d, e Immunofluorescence (IF) with anti-paxillin antibodies and 4′,6-diamidino-2-phenylindole (DAPI) staining of SAMHD1 knockdown SN12C cells or SAMHD1 overexpressed Caki-1 cells. Scale bar, 20 μm. f mCherry-cortactin was transfected as a control, and SAMHD1 was knocked down in SN12C cells for lamellipodium observation (left). Scale bar, 20 μm. Graphs present the lamellipodium area in each cell (right) (n = 20 cells). Data are presented as the mean ± SEM. ***p < 0.001. g SAMHD1-overexpressing Caki-1 cells and control cells were transfected with mCherry-cortactin for lamellipodium observation (left). Scale bar, 10 μm. Graphs present the lamellipodium area in each cell (right) (n = 20 cells). Data are presented as the mean ± SEM. ***p < 0.001.
SAMHD1 increases small GTPase activities, especially Rac1 activity
To further investigate the molecular mechanism that regulates the SAMHD1-mediated migration of ccRCC cells, we analyzed the molecular function category from the GSEA and found that the subcategories were mainly related to proteins regulating cell migration (Fig. 5a). In particular, the subcategories of small GTPase protein binding and Rac GTPase binding, which are essential for initiating lamellipodia formation, had high enrichment scores (Fig. 5b, c). To examine the causality between small GTPase activities and SAMHD1, Rac1 and cdc42 activities were measured following SAMHD1 knockdown or overexpression. Rac1 activity was decreased by SAMHD1 knockdown; however, increasing levels of exogenous SAMHD1 increased Rac1 activity to a similar level as that in the control (Fig. 5d). The activation of Rac1 and cdc42 in the early stages of cell migration is well established39. Consistent with the findings in previous reports, cdc42 activity with SAMHD1 knockdown and overexpression showed similar patterns as Rac1 activity (Fig. 5e). Rho A activity was also measured, but no significant difference in activity was observed (data not shown). Furthermore, to determine whether SAMHD1-mediated ccRCC cell migration is dependent on GTPase activation, a wound-healing assay was performed using small GTPase inhibitor treatments. Among the small GTPase inhibitors, the Rac1 inhibitor inhibited ccRCC cell migration to the same level as SAMHD1 knockdown (Fig. 5f). Similar results were observed in cells overexpressing SAMHD1, the ability of SAMHD1 to promote cell migration was completely blocked by the Rac1 inhibitor, NSC23766 (Fig. 5g). Our results suggest that Rac1 activation is essential for SAMHD1-induced cell migration.

a GO analysis was performed, and subcategories in the molecular function category were highly correlated with the high expression of SAMHD1. b, c Subcategories of the molecular function category related to cell migration. **p < 0.01. d A G-LISA-based assay was performed to measure GTP-bound Rac1 activity in SAMHD1-knockdown SN12C cells and SAMHD1-overexpressing Caki-1 cells. Data are presented as the mean ± SD. The data shown are representative of three independent experiments. **p < 0.01. e GTP-bound-cdc42 activity in SAMHD1-knockdown SN12C cells and SAMHD1-overexpressed Caki-1 cells. Data are presented as the mean ± SD. The experiments were independently performed three times. *p < 0.05; ***p < 0.001. f Wound-healing assay of cell migration ability with FAK inhibitor (PF573228), Rho A inhibitor (fasudil), Rac1 inhibitor (NSC23766), and cdc42 inhibitor (ML141). SAMHD1-knockdown SN12C cells were treated with each corresponding inhibitor at 10 μM over 24 h. The relative wound-healing area was normalized to that of the controls. Data are presented as the mean ± SD. The data shown are representative of three independent experiments. ***p < 0.001. g A wound healing assay was performed as in (f) in SAMHD1-overexpressing Caki-1 cells. The relative wound-healing area was normalized to that of DMSO-treated controls. *p < 0.05; ***p < 0.001. Data are presented as the mean ± SD of three independent experiments.
SAMHD1 binds with cortactin to promote endocytosis
Endocytosis is an important recycling system during cancer progression, especially during metastasis. Cancer cells recycle cell-surface proteins for cell migration by internalizing selective vesicles. Hence, the faster endocytosis occurs, the faster metastasis proceeds40,41. Notably, GSEA revealed a relationship between SAMHD1 expression and early endosomes as well as recycling endosomes (Fig. 6a, b). Since the initiation of endocytosis stimulates Rac1 activation42, we hypothesized that endocytosis might be involved in the SAMHD1-mediated migration of ccRCC cells. To investigate whether the formation of intracellular vesicles is regulated by SAMHD1, the expression of EEA1, an early endosome marker, was analyzed using confocal microscopy. SAMHD1 knockdown reduced the total number of EEA1-expressing vesicles in SN12C cells (Fig. 6c). In contrast, overexpression of SAMHD1 increased the number of EEA1-expressing vesicles by two-fold compared to that in the Caki-1 control cells (Fig. 6d). These results indicate that SAMHD1 promotes endosome formation.

a, b GSEA analysis of SAMHD1 mRNA levels and endocytosis-related signaling pathways. c ACHN cells were transfected with control or siSAMHD1 and immunolabeled for EEA1. The nuclei were stained with DAPI. Scale bar, 20 μm. Graphs present the number of EEA1-expressing vesicles per single cell (n = 55 cells). Data are presented as the mean ± SEM. The experiments were independently performed three times. ***p < 0.001. d SAMHD1-overexpressing Caki-1 cells were immunolabeled for EEA1. The number of EEA1-expressing vesicles per single cell is shown (n = 55 cells). Scale bar, 20 μm. Data are presented as the mean ± SEM. The data shown are representative of three independent experiments. ***p < 0.001. e SAMHD1 colocalized with cortactin in the cytoplasmic circular-shaped organelles and on the lamellipodia. f Exogenous Flag-tagged SAMHD1 was immunoprecipitated with magnetic Flag beads from Flag-SAMHD1-overexpressing Caki-1 cells. g Endogenous SAMHD1 was immunoprecipitated from SAMHD1-knockdown SN12C cells. Anti-cortactin antibodies were used to examine SAMHD1 and cortactin binding. IgGs were incubated with cell lysates as negative controls. h Endogenous cortactin was immunoprecipitated from SAMHD1-downregulated ACHN cells. Anti-SAMHD1 antibody was used to examine SAMHD1 and cortactin binding. IgG was incubated with cell lysates as a negative control. i, j Phosphorylated cortactin (Y421) and total cortactin levels were evaluated using western blotting in SAMHD1-knockdown ACHN and SN12C cells or SAMHD1-overexpressed Caki-1 and Caki-2 cells. k IHC analysis of SAMHD1 and phosphorylated cortactin (Y421) in ccRCC patient tissues. Scale bar, 50 μm. l Areas with positive IHC staining were measured and are shown on the graph. Data are presented as the mean ± SD. ***p < 0.001; **p < 0.01. m Correlation between SAMHD1 and phosphorylated cortactin (Y421) expression from IHC analysis of ccRCC patient tissues.
Consistent with previous reports, SAMHD1 was mainly expressed in the nucleus. However, SAMHD1 was observed in the cytoplasm and cell membrane to a lesser extent, but SAMHD1 localization in the cytoplasm and cell membrane was rarely found in SAMHD1-knockdown cells (Supplementary Fig. 3a). In addition, Flag-tagged exogenous SAMHD1 was found not only in the nucleus but also in the cytoplasm and cell membrane (Supplementary Fig. 3b). Interestingly, we observed that SAMHD1 colocalized with cortactin in the lamellipodia and cytoplasm (Fig. 6e). Based on this observation, we examined the interaction between SAMHD1 and cortactin using immunoprecipitation and observed exogenous and endogenous binding of SAMHD1 and cortactin (Fig. 6f–h). Tyrosine phosphorylation of cortactin is important for endosomal vesicle formation27,43,44. We found that the level of cortactin tyrosine phosphorylation was decreased in SAMHD1-depleted ACHN and SN12C cells (Fig. 6i). Conversely, SAMHD1 overexpression in Caki-1 and Caki-2 cells potentiated cortactin tyrosine phosphorylation levels (Fig. 6j). For further confirmation, we analyzed the correlation between SAMHD1 and cortactin phosphorylation in paired cancer and normal tissues from patients with ccRCC. Both SAMHD1 and phosphorylated cortactin levels were higher in cancer tissues than in adjacent normal tissues (Fig. 6k, l). The phosphorylation level of cortactin was positively correlated with the expression of SAMHD1 in tumor tissues (Fig. 6m).
To further determine the impact of endocytosis on SAMHD1-induced endosome formation, we treated cells with Dynasore, an endocytosis inhibitor. Dynasore treatment attenuated the colocalization of SAMHD1 and EEA1 in endosomal vesicles (Fig. 7a, b). In addition, the binding of the endosomal formation complex with SAMHD1 was markedly decreased by the inhibition of endocytosis (Fig. 7c). Dynasore also attenuated cortactin phosphorylation in SAMHD1-overexpressing cells (Fig. 7d), suggesting that cortactin phosphorylation is involved in SAMHD1-mediated endosomal complex formation.

a IF staining with an anti-SAMHD1 antibody, an anti-EEA1 antibody, and DAPI in SAMHD1-overexpressed Caki-1 cells treated with 80 μM Dynasore over 24 h. b The number of EEA1 vesicles colocalized with SAMHD1 per cell was quantified and is shown on the graph (n = 30 cells). Data are presented as the mean ± SD. The data shown are representative of three independent experiments. ***p < 0.001. c IP assay was performed using Flag-tagged beads in Flag-SAMHD1-overexpressing Caki-1 cell lysates treated with 80 μM Dynasore over 24 h. d Phosphorylated cortactin (Y421) and total cortactin expression in SAMHD1-overexpressed Caki-1 cells treated with 80 μM Dynasore over 24 h and analyzed using western blotting. e, f Wound-healing assay of cell migration ability in SAMHD1-knockdown SN12C cells or in SAMHD1-overexpressed Caki-1 cells treated with 80 μM Dynasore. The relative wound-healing area was normalized to that of DMSO-treated controls. Data are presented as the mean ± SD. The data shown are representative of three independent experiments. ***p < 0.001. g G-LISA-based assay of GTP-bound Rac1 activity in SAMHD1-overexpressing Caki-1 cells treated with 10 μM NSC23766 or 80 μM Dynasore over 24 h. ***p < 0.001. Data are presented as the mean ± SD of three independent experiments.
Furthermore, to determine whether SAMHD1-mediated ccRCC cell migration is maintained by endocytosis, we performed a wound-healing assay with an endocytosis inhibitor. As shown in Fig. 7e, the movement of cells was slowed down with Dynasore treatment in control cells compared with SAMHD1 knockdown cells. Furthermore, the ability of SAMHD1 to induce cell migration was eliminated after Dynasore treatment (Fig. 7f). Based on accumulating evidence that endocytosis contributes to Rac1 activation42,45,46, we investigated whether SAMHD1-induced endocytosis could regulate Rac1 activation in Dynasore-treated cells. Dynasore treatment impaired SAMHD1-mediated Rac1 activation (Fig. 7g), suggesting that Rac1 is downstream of endocytosis signaling. Collectively, these findings suggest that SAMHD1-mediated endocytosis promotes ccRCC cell migration through Rac1 activation.
SAMHD1 activates endosomal FAK signaling
FAK is a well-known binding partner of cortactin and participates in various cell migration processes, lamellipodia formation, and endocytosis. Autophosphorylation of FAK at Y397 is necessary for cortactin phosphorylation and Rac1 activation22,47,48. Accordingly, we examined whether FAK phosphorylation was regulated by SAMHD1. FAK phosphorylation levels at Y397 decreased in SAMHD1-knockdown cells (Fig. 8a); in contrast, SAMHD1 overexpression increased FAK phosphorylation levels (Fig. 8b). Dynasore treatment impaired SAMHD1-induced FAK phosphorylation, indicating that SAMHD1-mediated endocytosis is required for FAK phosphorylation (Fig. 8c). To determine whether FAK participates in SAMHD1-induced endosome formation, we treated cells with the selective FAK inhibitor PF573228. In the presence of PF573228, SAMHD1-overexpressing cells did not form EEA1-expressing vesicles (Fig. 8d, e). In addition, treatment with PF573228 inhibited the binding of endosomal components to SAMHD1 (Fig. 8f). Consistent with endosomal complex formation, SAMHD1-induced cortactin phosphorylation was also diminished by FAK inhibitor treatment (Fig. 8g). To investigate whether FAK signaling is essential for SAMHD1-mediated ccRCC cell migration, we performed a wound-healing assay with FAK inhibitor treatment. PF573228 treatment reduced the motility of control cells to a level similar to that of SAMHD1-deficient cells (Fig. 8h). In addition, the effect of SAMHD1 overexpression on cell migration was reduced by PF573228 treatment (Fig. 8i). Collectively, our results suggest that SAMHD1-induced endosomal FAK signaling promotes ccRCC cell migration.

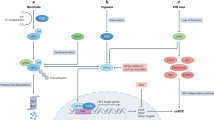
a, b Western blot analysis of phospho-FAK (Y397) and total FAK levels in SAMHD1-knockdown ACHN and SN12C cells or SAMHD1-overexpressed Caki-1 and Caki-2 cells . c Western blot analysis of phosphorylated FAK (Y397) and total FAK expression in SAMHD1-overexpressed Caki-1 cells treated with 80 μM Dynasore over 24 h. d SAMHD1-overexpressed Caki-1 cells were treated with 10 μM PF573228 for 24 h and immunolabeled for EEA1. The nuclei were stained with DAPI. Scale bar, 20 μm. e Graphs show the number of EEA1 vesicles per single cell (n = 55 cells). ***p < 0.001. f IP assay was performed using Flag-tagged beads in Flag-SAMHD1-overexpressing Caki-1 cell lysates treated with 10 μM PF573228 over 24 h. g Western blot analysis of phosphorylated FAK (Y397) and phosphorylated cortactin (Y421) expression in SAMHD1-overexpressed Caki-1 cells treated with 10 μM PF573228 over 24 h. h, i Wound-healing assay of cell migration ability in SAMHD1-knockdown SN12C cells or SAMHD1-overexpressed Caki-1 cells treated with 10 μM PF573228 . The relative wound-healing area was normalized to that of the controls. Data are presented as the mean ± SEM. The experiments were performed at least three times. *p < 0.05, ***p < 0.001. j IHC of SAMHD1 and p-FAK (Y397) expression in tissues of patients with ccRCC. Scale bar, 50 μm. k Areas with positive IHC staining were measured and are presented in the graph. Data are presented as the mean ± SD. *p < 0.05. l SAMHD1 promotes ccRCC metastasis through endosomal FAK signaling activation.
Finally, we analyzed FAK activation in tumor tissues from patients with ccRCC. Consistent with the pattern of SAMHD1 expression, FAK phosphorylation levels were higher in cancer tissues than in normal tissues (Fig. 8j, k). These findings confirm the important role of SAMHD1-promoted FAK phosphorylation in mediating ccRCC development.
Discussion
Every year, 403 000 new cases of renal cell carcinoma are diagnosed worldwide. Most patients with renal cancer only become aware of symptoms after the cancer has progressed to a later stage, with only 10% of patients reporting symptoms in the early stages of the disease. Therefore, 33% of patients are diagnosed with stage IV metastatic renal cell carcinoma, which has a 5-year survival rate of only 12%49,50. Despite medical and therapeutic advances, effective strategies to prevent renal cancer metastasis are limited; therefore, insights into the molecular mechanisms promoting kidney cancer metastasis are urgently needed.
In the present study, we identified SAMHD1 as an oncogene in ccRCC and proposed a novel mechanism in cell migration. We found that SAMHD1-mediated endocytic FAK signaling promotes lamellipodia formation in ccRCC cells (Fig. 8l), SAMHD1 binds to cortactin to promote early endosome formation and stimulates Rac1 activation via FAK autophosphorylation, and activated Rac1 promotes lamellipodia protrusion, which initiates ccRCC cell migration.
The function of SAMHD1 in cancer is unclear; whether SAMHD1 plays the role of an oncogene or cancer suppressor remains controversial. We assumed that SAMHD1 might function through multiple pathways in different types of cancer. Wu et al. 51 reported that SAMHD1 acts as a tumor suppressor through the STING pathway during cancer progression in lung adenocarcinoma. TCGA analysis showed that SAMHD1 mRNA levels were much lower in tumor tissues than in nontumor tissues in several cancer types, such as colorectal adenocarcinoma, bladder urothelial carcinoma, lung adenocarcinoma, and lung squamous cell carcinoma. In contrast, other types of cancers, such as glioma, uterine corpus endometrial carcinoma, and especially pankidney cancer, had much greater SAMHD1 mRNA levels in tumor tissues than in nontumor tissues (Fig. 1a), indicating that SAMHD1 serves as a tumor suppressor or oncogene depending on the type of cancer. Herein, we provide strong evidence for the oncogenic roles of SAMHD1 in ccRCC supported by various in vitro assays together with ccRCC patient sample analysis. Our findings suggest a new clinical approach with SAMHD1 as a therapeutic marker for ccRCC treatment.
SAMHD1 expression was significantly increased upregulated in cancer tissues compared to normal tissues in RCC among several cancer types. The overexpression of SAMHD1 in RCC tumor tissues was also related to worse survival prognoses in groups with high SAMHD1 expression, especially for patients with the ccRCC subtype. Although we found no significant differences in survival between high and low SAMHD1-expression groups in PRCC and chRCC, some patients expressing high SAMHD1 showed better survival rates than those of patients expressing low SAMHD1, although this finding may be an artifact of small sample size52. Only 20% of RCC patients are diagnosed with the PRCC and chRCC subtypes49; therefore, less information is available for PRCC and chRCC than for ccRCC. This is especially the case for chRCC datasets, which lack the appropriate sample size and longitudinal data for survival analysis.
SAMHD1 is mainly observed in the nucleus and is thus widely considered to be a nuclear protein8,53; however, SAMHD1 has also been detected in the cytoplasm. Recent reports have indicated the importance of nucleocytoplasmic shuttling to the function of SAMHD1. SAMHD1 can translocate into the cytoplasm through stimulation by growth factors, and shifting its localization to the cytoplasm is required for LINE-1 inverse enzyme inhibition9,54. In contrast to studies stating that SAMHD1 only has catalytic activity as a deoxyribonucleoside triphosphohydrolase in the nucleus8,53, the restriction activity of HIV-I was confirmed in an NLS mutant of SAMHD1 or with its NLS inactivated cells, suggesting a cytoplasmic function for SAMHD1. In this study, we confirmed that SAMHD1 was localized in both the cytoplasm and cell membrane in addition to the nucleus. Furthermore, we found that SAMHD1 participates in lamellipodia formation and endocytosis by binding to cortactin. This result is consistent with the findings of a previous study that demonstrated that SAMHD1 binds to the transmembrane protein CD8155. The function of SAMHD1 is regulated via various posttranslational modifications, such as phosphorylation, acetylation, methylation, and SUMOylation16,56,57,58,59. In particular, SAMHD1 phosphorylation at T592 is important for determining its localization in the cytoplasm60. Therefore, we investigated whether the level of SAMHD1 phosphorylated at T592 affected patient prognosis but found no significant difference in the phosphorylation level of SAMHD1 normalized to total SAMHD1 between the normal and tumor tissues of ccRCC patients under various pathological conditions (data not shown). Although SAMHD1 phosphorylation at T592 is not involved in regulating the malignancy of ccRCC, further studies are required to examine whether the ability of SAMHD1 to induce endocytosis and lamellipodia formation are regulated by other posttranslational modifications.
We found that SAMHD1 colocalized with EEA1, which led to the formation of early endosomes to facilitate cell migration (Fig. 7a). Consistent with our findings, Rocha-Perugini et al. 55 reported that cytoplasmic vesicles generated by SAMHD1 are surrounded by EEA1, which protects them from proteasomal degradation. They also showed that vesicles containing SAMHD1 were unrelated to late endosomes, multivesicular bodies (CD63), or lysosomes. Yuan et al. 61 also showed that SAMHD1 was a predicted binding partner of Rab21 in Rab21-mediated vesicle trafficking using mass spectrometry. Increasing evidence suggests that Rab21 is involved in early endosome formation and is constantly recycled, together with vesicles, to the cell membrane to promote cell migration rather than degradation31,62,63,64. Based on these previous findings, our study provides new insights into the regulatory mechanisms of SAMHD1-mediated early endosome formation.
Cortactin is an actin-binding protein that interacts with a wide range of proteins and participates in lamellipodia development in cell adhesion and migration23,24,27. We identified SAMHD1 as a novel binding partner of cortactin and confirmed that they were colocalized in the plasma membrane and endosomes (Fig. 6e). Although the exact binding site of these proteins is unclear, GSEA analysis suggests that SAMHD1 binds to the SH3 domain, one of three major cortactin domains, with a high probability (Fig. 5a). It is likely that SAMHD1 binds to the SH3 domain of cortactin as this domain is where other binding partners interact with cortactin. Further studies are needed to determine the cortactin binding domain that SAMHD1 interacts with.
Overall, our study presents a clearly elucidated pathway by which SAMHD1 regulates endosomal FAK signaling. In this cascade signaling axis, we identified cortactin as a novel binding partner of SAMHD1 that participates in endocytosis and lamellipodia formation. Furthermore, activated FAK signaling stimulates Rac1 activity, inducing lamellipodia protrusion for ccRCC cell migration. In conclusion, the SAMHD1-mediated endosomal FAK signaling axis could serve as a novel therapeutic target for ccRCC.
References
Rice, G. I. et al. Mutations involved in Aicardi-Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nat. Genet. 41, 829–832 (2009).
Kim, E. T., White, T. E., Brandariz-Núñez, A., Diaz-Griffero, F. & Weitzman, M. D. SAMHD1 restricts herpes simplex virus 1 in macrophages by limiting DNA replication. J. Virol. 87, 12949–12956 (2013).
Goldstone, D. C. et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 480, 379–382 (2011).
Jeong, G. U., Park, I. H., Ahn, K. & Ahn, B. Y. Inhibition of hepatitis B virus replication by a dNTPase-dependent function of the host restriction factor SAMHD1. Virology 495, 71–78 (2016).
Brandariz-Nuñez, A. et al. Role of SAMHD1 nuclear localization in restriction of HIV-1 and SIVmac. Retrovirology 9, 49 (2012).
Coquel, F. et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 557, 57–61 (2018).
Schaller, T., Pollpeter, D., Apolonia, L., Goujon, C. & Malim, M. H. Nuclear import of SAMHD1 is mediated by a classical karyopherin α/β1 dependent pathway and confers sensitivity to VpxMAC induced ubiquitination and proteasomal degradation. Retrovirology 11, 29 (2014).
Hofmann, H. et al. The Vpx lentiviral accessory protein targets SAMHD1 for degradation in the nucleus. J. Virol. 86, 12552–12560 (2012).
Mauney, C. H. et al. The SAMHD1 dNTP triphosphohydrolase Is controlled by a redox switch. Antioxid. Redox Signal. 27, 1317–1331 (2017).
Schott, K. et al. SAMHD1 in cancer: curse or cure? J. Mol. Med. (Berl). 100, 351–372 (2022).
Rentoft, M. et al. Heterozygous colon cancer-associated mutations of SAMHD1 have functional significance. Proc. Natl. Acad. Sci. 113, 4723–4728 (2016).
Wang, T. et al. SAMHD1 mutations and expression in mantle cell lymphoma patients. Front. Oncol. 11, 763151 (2021).
Herold, N. et al. SAMHD1 protects cancer cells from various nucleoside-based antimetabolites. Cell Cycle. 16, 1029–1038 (2017).
Rassidakis, G. Z. et al. Low-level expression of SAMHD1 in acute myeloid leukemia (AML) blasts correlates with improved outcome upon consolidation chemotherapy with high-dose cytarabine-based regimens. Blood Cancer J. 8, 98 (2018).
Bühler, M. M. et al. SAMHD1 mutations in mantle cell lymphoma are recurrent and confer in vitro resistance to nucleoside analogues. Leuk. Res. 107, 106608 (2021).
Wang, J. L., Lu, F. Z., Shen, X. Y., Wu, Y. & Zhao, L. T. SAMHD1 is down regulated in lung cancer by methylation and inhibits tumor cell proliferation. Biochem. Biophys. Res. Commun. 455, 229–233 (2014).
Sheetz, M. P. Cell control by membrane-cytoskeleton adhesion. Nat. Rev. Mol. Cell Biol. 2, 392–396 (2001).
McMahon, H. T. & Gallop, J. L. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature 438, 590–596 (2005).
Ridley, A. J. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 16, 522–529 (2006).
Brakebusch, C. & Fässler, R. The integrin-actin connection, an eternal love affair. EMBO J. 22, 2324–2333 (2003).
Shi, Q. & Boettiger, D. A novel mode for integrin-mediated signaling: tethering is required for phosphorylation of FAK Y397. Mol. Biol. Cell. 14, 4306–4315 (2003).
Chang, F., Lemmon, C. A., Park, D. & Romer, L. H. FAK potentiates Rac1 activation and localization to matrix adhesion sites: a role for betaPIX. Mol. Biol. Cell. 18, 253–264 (2007).
Weed, S. A., Du, Y. & Parsons, J. T. Translocation of cortactin to the cell periphery is mediated by the small GTPase Rac1. J. Cell Sci. 111, 2433–2443 (1998).
Bryce, N. S. et al. Cortactin promotes cell motility by enhancing lamellipodial persistence. Curr. Biol. 15, 1276–1285 (2005).
Chrzanowska-Wodnicka, M. & Burridge, K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J. Cell Biol. 133, 1403–1415 (1996).
Burridge, K. & Wennerberg, K. Rho and Rac take center stage. Cell 116, 167–179 (2004).
Kruchten, A. E., Krueger, E. W., Wang, Y. & McNiven, M. A. Distinct phospho-forms of cortactin differentially regulate actin polymerization and focal adhesions. Am. J. Physiol. Cell Physiol. 295, C1113–C1122 (2008).
Wang, W., Liu, Y. & Liao, K. Tyrosine phosphorylation of cortactin by the FAK-Src complex at focal adhesions regulates cell motility. BMC Cell Biol. 12, 49 (2011).
Hamidi, H. & Ivaska, J. Every step of the way: integrins in cancer progression and metastasis. Nat. Rev. Cancer. 18, 533–548 (2018).
Fletcher, S. J. & Rappoport, J. Z. Moving forward: polarised trafficking in cell migration. Trends Cell Biol. 20, 71–78 (2010).
Roberts, M., Barry, S., Woods, A., van der Sluijs, P. & Norman, J. PDGF-regulated rab4-dependent recycling of alphavbeta3 integrin from early endosomes is necessary for cell adhesion and spreading. Curr. Biol. 11, 1392–1402 (2001).
Bhuin, T. & Roy, J. K. Rab proteins: the key regulators of intracellular vesicle transport. Exp. Cell Res. 328, 1–19 (2014).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. 102, 15545–15550 (2005).
Hwang, J. S., Cha, E. H., Park, B., Ha, E. & Seo, J. H. PBN inhibits a detrimental effect of methamphetamine on brain endothelial cells by alleviating the generation of reactive oxygen species. Arch. Pharm. Res. 43, 1347–1355 (2020).
Barzegar, M. et al. SKLB188 inhibits the growth of head and neck squamous cell carcinoma by suppressing EGFR signalling. Br. J. Cancer. 117, 1154–1163 (2017).
Moch, H., Cubilla, A. L., Humphrey, P. A., Reuter, V. E. & Ulbright, T. M. The 2016 WHO classification of tumours of the urinary system and male genital organs-Part A: Renal, penile, and testicular tumours. Eur. Urol. 70, 93–105 (2016).
Dongre, A. & Weinberg, R. A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 20, 69–84 (2019).
Webb, D. J. et al. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 6, 154–161 (2004).
Ridley, A. J. Life at the leading edge. Cell 145, 1012–1022 (2011).
Paul, N. R., Jacquemet, G. & Caswell, P. T. Endocytic trafficking of integrins in cell migration. Curr. Biol. 25, R1092–R1105 (2015).
Arriagada, C. et al. Focal adhesion kinase-dependent activation of the early endocytic protein Rab5 is associated with cell migration. J. Biol. Chem. 294, 12836–12845 (2019).
Díaz, J. et al. Rab5 is required in metastatic cancer cells for Caveolin-1-enhanced Rac1 activation, migration and invasion. J. Cell Sci. 127, 2401–2406 (2014).
Luo, C. et al. CXCL12 induces tyrosine phosphorylation of cortactin, which plays a role in CXC chemokine receptor 4-mediated extracellular signal-regulated kinase activation and chemotaxis. J. Biol. Chem. 281, 30081–30093 (2006).
Rossé, C. et al. Control of MT1-MMP transport by atypical PKC during breast-cancer progression. Proc. Natl. Acad. Sci. 111, E1872–E1879 (2014).
Ménard, L., Parker, P. J. & Kermorgant, S. Receptor tyrosine kinase c-Met controls the cytoskeleton from different endosomes via different pathways. Nat. Commun. 5, 3907 (2014).
Joffre, C. et al. A direct role for Met endocytosis in tumorigenesis. Nat. Cell Biol. 13, 827–837 (2011).
Tomar, A., Lawson, C., Ghassemian, M. & Schlaepfer, D. D. Cortactin as a target for FAK in the regulation of focal adhesion dynamics. PLoS One. 7, e44041 (2012).
Alanko, J. et al. Integrin endosomal signalling suppresses anoikis. Nat. Cell Biol. 17, 1412–1421 (2015).
Padala, S. A. et al. Epidemiology of renal cell carcinoma. World J. Oncol. 11, 79–87 (2020).
Ferlay, J. et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer. 144, 1941–1953 (2019).
Wu, Y. et al. SAMHD1 can suppress lung adenocarcinoma progression through the negative regulation of STING. J. Thorac. Dis. 13, 189–201 (2021).
Liu, J. et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 173, 400–416.e411 (2018).
Guo, H. et al. Identification of critical regions in human SAMHD1 required for nuclear localization and Vpx-mediated degradation. PLoS One. 8, e66201 (2013).
Du, J. et al. Nucleocytoplasmic shuttling of SAMHD1 is important for LINE-1 suppression. Biochem. Biophys. Res. Commun. 510, 551–557 (2019).
Rocha-Perugini, V. et al. CD81 association with SAMHD1 enhances HIV-1 reverse transcription by increasing dNTP levels. Nat. Microbiol. 2, 1513–1522 (2017).
Lee, E. J. et al. SAMHD1 acetylation enhances its deoxynucleotide triphosphohydrolase activity and promotes cancer cell proliferation. Oncotarget 8, 68517–68529 (2017).
Welbourn, S., Dutta, S. M., Semmes, O. J. & Strebel, K. Restriction of virus infection but not catalytic dNTPase activity is regulated by phosphorylation of SAMHD1. J. Virol. 87, 11516–11524 (2013).
Silva, S. et al. Promoter methylation regulates SAMHD1 gene expression in human CD4+ T cells. J. Biol. Chem. 288, 9284–9292 (2013).
Martinat, C. et al. SUMOylation of SAMHD1 at lysine 595 is required for HIV-1 restriction in non-cycling cells. Nat. Commun. 12, 4582 (2021).
Batalis, S. et al. SAMHD1 phosphorylation at T592 regulates cellular localization and S-phase progression. Front. Mol. Biosci. 8, 724870 (2021).
Yuan, Q. et al. PKN1 directs polarized RAB21 vesicle trafficking via RPH3A and Is important for neutrophil adhesion and ischemia-reperfusion Injury. Cell Rep. 19, 2586–2597 (2017).
Pellinen, T. et al. Small GTPase Rab21 regulates cell adhesion and controls endosomal traffic of beta1-integrins. J. Cell Biol 173, 767–780 (2006).
Caswell, P. T., Vadrevu, S. & Norman, J. C. Integrins: masters and slaves of endocytic transport. Nat. Rev. Mol. Cell Biol. 10, 843–853 (2009).
Roberts, M. S., Woods, A. J., Dale, T. C., Van Der Sluijs, P. & Norman, J. C. Protein kinase B/Akt acts via glycogen synthase kinase 3 to regulate recycling of alpha v beta 3 and alpha 5 beta 1 integrins. Mol. Cell. Biol. 24, 1505–1515 (2004).
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2016R1A6A1A03011325 to J.H.S.), the Korean government (MSIT) (NRF-2021R1A2B5B01002223 to K.-W.K.) and the KBRI (Korea Brain Research Institute) basic research program through the KBRI funded by the Korean Ministry of Science and ICT (22-BR-05-03 to T.S.)
Author information
Authors and Affiliations
Contributions
Conceptualization, S.A. and J.H.S.; Methodology, S.A.; Validation, Y.-J.S.; Formal analysis, S.A., and T.S.; Investigation, S.A, T.T.L.V., H.C., J.L., and E.H.; Resources, J.K., B.H.K., M.C.; Writing, S.A. and J.H.S.; Visualization, S.A.; Supervision, Y.-J.S, K.-W.K., and J.H.S.; Project Administration, J.H.S; Funding Acquisition, K.-W.K. and J.H.S.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
An, S., Vo, T.T.L., Son, T. et al. SAMHD1-induced endosomal FAK signaling promotes human renal clear cell carcinoma metastasis by activating Rac1-mediated lamellipodia protrusion. Exp Mol Med 55, 779–793 (2023). https://doi.org/10.1038/s12276-023-00961-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-023-00961-x
This article is cited by
-
Fumigaclavine C ameliorates liver steatosis by attenuating hepatic de novo lipogenesis via modulation of the RhoA/ROCK signaling pathway
BMC Complementary Medicine and Therapies (2023)
