
Abstract
Comprehensive molecular analyses revealed that papillary renal cell carcinoma (PRCC) is a heterogenous entity. Papillary renal neoplasm with reverse polarity (PRNRP) is a subset of PRCC with characteristic histomorphologies such as low-grade nuclear features, inverted nuclear location, eosinophilic cytoplasm, and indolent clinical behavior. We tried to define the molecular, clinicopathological, histologic, and immunohistochemical features of PRNRP by comparing them with type 1 PRCC (PRCC1) and type 2 PRCC (PRCC2). A cohort of 30 PRNRP, 23 PRCC1, and 26 PRCC2 cases was used. Targeted sequencing of 90 cancer-related genes including KRAS was performed in 26 PRNRP tumor samples. PNA-mediated clamping PCR of KRAS was performed using paired normal and tumor DNA from 30 PRNRP, 23 PRCC1, and 26 PRCC2 cases. Tissue microarray slides were made in three cores per tumor, which were stained with cytokeratin 7 (CK7), alpha-methylacyl-CoA racemase (AMACR), epithelial membrane antigen (EMA), E-cadherin, vimentin, and CD10. Recurrent mutations in KRAS were detected in 28 of the 30 PRNRPs. However, there were no KRAS mutations in any PRCC1 or PRCC2 cases. PRNRP exhibited distinct clinicopathological features: small tumor size, lower pathologic T stage, and no disease-specific death during the follow-up period. Histologically, peritumoral lymphoid aggregation, prominent papillary architecture (>80% of tumor), hyalinized papillae, inverted nuclear location, and lower nuclear grade were observed. PRNRP was usually positive for CK7, AMACR, EMA, and E-cadherin, and negative for CD10. The findings suggest that PRNRP is a subtype of papillary renal neoplasm that is different from PRCC1 or PRCC2 in terms of molecular, clinicopathological, histological, and immunohistochemical features.
Similar content being viewed by others

Introduction
Papillary renal cell carcinoma (PRCC) is the second most common morphologic type of renal cell carcinoma [1]. PRCC cases are classified as type 1 or type 2 based on histological findings. Type 1 PRCC (PRCC1) shows papillae lined by single-layered cells with scant pale cytoplasm, whereas papillae in type 2 PRCC (PRCC2) are lined by cells having abundant eosinophilic cytoplasm and frequently higher nucleolar grades [2]. However, there have been some PRCC cases that are neither PRCC1 nor PRCC2 [3]. Recently, the cancer genome atlas (TCGA) study revealed that PRCC2 is a heterogeneous group composed of at least three molecular subtypes; some cases exhibit more aggressive biologic behavior, and others an indolent prognosis [4, 5].
Papillary renal tumors with an oncocytic cytoplasm can be found in oncocytic PRCC, PRCC2, hereditary leiomyomatosis and renal cell carcinoma (HLRCC)-associated RCC, MiT family (MITF) translocation RCC, and acquired cystic disease-associated RCC. Oncocytic PRCC is a subtype of PRCC, composed of cells with “voluminous, fully granular, evenly distributed eosinophilic cytoplasm and oncocytoma-like (usually with low nucleolar grade) nuclei” [6]. However, since oncocytic PRCC has been used to refer to PRCC with abundant eosinophilic cytoplasms, this term may include any PRCCs with oncocytic cytoplasm, regardless of nuclear morphology [7,8,9,10]. Among them, a subset of tumors showed distinct histomorphological and clinical features, which are low-grade nuclear features, linear nuclear arrangement apart from the base, eosinophilic cytoplasm, and indolent clinical course. These tumors have been reported as oncocytic papillary renal cell carcinoma [11], adult papillary renal tumor with oncocytic cells [7], papillary renal cell carcinoma with oncocytic cells and nonoverlapping low-grade nuclei [8], oncocytic papillary renal cell carcinoma with inverted nuclear pattern [12], and papillary renal neoplasm with reverse polarity (PRNRP) [13].
Since the term “oncocytic papillary renal cell carcinoma” does not fit the clinicopathological features of this disease, we consider the term “PRNRP” appropriate for this subgroup of oncocytic PRCC [13].
In this study, we tried to find the molecular, clinicopathological, histomorphological, and immunohistochemical characteristics of PRNRP by comparing it with those of PRCC1 and PRCC2.
Materials and methods
Case selection
Thirty cases of PRNRP, 23 PRCC1 and 26 PRCC2, were included in this study. They were obtained between January 2001 and December 2017 from six university hospitals in the Republic of Korea (Chonnam National University Hospital, Chonnam National University Hwasun Hospital, Asan Medical Center, Chonbuk National University Hospital, Chosun University Hospital, Wonkwang University Hospital). Two pathologists (Kim and Choi) independently reviewed the hematoxylin- and eosin-stained slides in blind fashion. When the diagnoses of the two pathologists were agreed upon, they were included in the study. In cases in which the diagnoses were not agreed upon, both of the pathologists discussed and decided whether to include or exclude. PRCC1 and PRCC2 are defined as described in the WHO classification of tumors of the urinary system and male genital organs [6]. PRNRP is characterized with papillary or tubule-papillary renal neoplasm(s) with low-grade nuclei, inverted nuclear location, and/or abundant and eosinophilic cytoplasm. However, not all of the PRNRP satisfied all of the above characteristics. In those case, decision was made after consideration of the mmunohistochemical findings. Papillary tumors with large nuclei with prominent inclusion-like eosinophilic nucleoli were excluded in order to rule out HLRCC-associated RCCs. Cases with dual cytoplasmic tones, biphasic TFEB tranlocation RCC-like morphology, and nuclear pseudoinclusion were excluded in order to rule out RCC with MiT aberration [14]. In addition, cases with oxalate crystal deposition, cribriform/sieve-like architecture, and cystic background of adjacent non-neoplastic tissue were eliminated in order not to include acquired cystic disease-associated RCC. The clinical information including follow-up data was collected from medical records. This study was approved by the Institutional Review Board of Chonnam National University Hwasun Hospital (CNUHH-2019-006).
DNA extraction and targeted sequencing
The targeted sequencing of tumor DNA was performed in 26 out of 30 PRNRP cases. The targeted sequencing was not performed in the remaining four cases, because their quality was not suitable for targeted sequencing. The tumor DNA was extracted from 10-μm macro-dissected slices of formalin-fixed paraffin-embedded tissue using a GeneRead™ DNA FFPE Kit (QIAGEN, Hilden, Germany). A targeted panel was used to capture the target regions of 90 cancer-related genes and all the coding exons of the 90 genes, to find single nucleotide variants (SNVs) and small insertions and deletions (InDels) in the following genes: AKT1, ABL1, ABL2, AKT2, AKT3, ALK, APC, AR, ARID1A, ARID1B, ARID2, ATM, ATR, ATRX, AURKA, AURKB, AXL, BCL2, BRAF, BRCA1, BRCA2, CCND1, CD274, CDH1, CDK4, CDK6, CDKN2A, CSF1R, CTNNB1, DDR2, EGFR, ERBB2, ERBB3, ERBB4, ESR1, FBXW7, FGFR1, FGFR2, FGFR3, FGFR4, FLT3, GNA11, GNAQ, GNAS, HRAS, IDH1, IDH2, IGF1R, JAK1, JAK2, JAK3, KDR, KIT, KRAS, MDM2, MET, MLH1, MPL, MSH2, MSH6, MTOR, MYC, MYCN, NF1, NF2, NOTCH1, NPM1, NRAS, NTRK1, NTRK2, NTRK3, PDGFRA, PDGFRB, PIK3CA, PIK3R1, POLE, PTEN, RB1, RET, ROS1, SMAD4, SMO, SRC, STK11, SYK, TOP1, TOP2A, TP53, TSC1, and VHL (SureSelect, Agilent Technologies, Santa Clara, CA, USA). The SureSelect targeted panel was prepared using 200 ng of genomic DNA as stated in the manufacturer's protocol. The genomic DNA was randomly fragmented by sonication (Covaris Inc., Woburn, MA, USA) followed by adapter ligation, purification, hybridization, and PCR analysis. Captured libraries were analyzed with an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, USA) and loaded onto the MiSeqDx instrument (Illumina, San Diego, USA) as recommended by the manufacturer.
Peptide nucleic acid (PNA)-mediated clamping PCR for detection of KRAS mutation
KRAS mutation status was analyzed in paired normal and tumor DNA of 30 PRNRP, 23 PRCC1, and 26 PRCC2 cases, respectively. DNA was extracted from 10-μm macro-dissected slices of paraffin blocks using the QIAamp DNA Mini Kit (QIAGEN). The assay was performed using the PNAClamp™ KRAS Mutation Detection Kit (Panagene, Daejeon, Korea) according to the manufacturer's protocol.
Bioinformatics analysis
Sequenced reads were aligned to the human reference genome (GRCh37 /hg19) with BWA-MEM (0.7.15) with default options [15]. To remove PCR duplicates from the aligned reads, the MarkDuplicates of the Picard package (https://broadinstitute.github.io/picard/) was used. De-duplicated reads were re-aligned at known InDels positions with the GATK (v2.3) IndelRealigner [16]. Base qualities were then re-calibrated using the GATK TableRecalibration. Somatic SNVs and short InDels were detected using VarDict and Mutect2 in GATK4 [17, 18]. Common and germline variants from somatic variant candidates were filtered out with common dbSNP (149 found in >1% of samples), The Genome Aggregation Database (gnomAD; r2.0.1), and the Korean Reference Genome Database (KRGDB, http://152.99.75.168/KRGDB/) [19, 20]. Final somatic variants were annotated using SnpEff (version 4.3i) [21]. False-positive variants were manually curated using the Integrative Genomics Viewer (IGV) [22]. The Bioconductor package GenVisR [23] was used to plot mutation lolliplots and waterfall plots. Since no paired normal tissue controls were available, the following filters were used: [1] the sequencing depth had at least 20 reads and a tumor variant allele frequency (VAF) ≥ 3%; [2] variant types (missense, frameshift, stop gain/loss, in-frame insertion/deletion, and protein-altering) were included; [3] VAF ≥ 10% or (3% ≤ VAF ≤ 10% and listed in the COSMIC database) [24]; (4) Mutect2 was run without a matched normal control; (5) variants found in more than 1% of total gnomAD or east-Asian gnomAD samples were excluded; and variants found in more than 1% of KRGDB normal controls were excluded.
Comparative analysis with TCGA dataset
Data of somatic mutations in renal cell carcinoma were downloaded and the digital images of the cases with KRAS mutations were reviewed in January 2019 from the cBioPortal repository [25, 26].
Tissue microarray (TMA) and immunohistochemistry
Three representative foci in each case were selected for TMA blocks. Three cores (2 mm in diameter) were obtained from each paraffin block to make a TMA block. Serial sections were prepared and stained with hematoxylin and eosin. Immunohistochemistry was carried out on the TMA slides using a Bond-Max automated IHC/ISH stainer (Leica Biosystems, Wetzlar, Germany) or a Ventana system (Benchmark Ultra, Tucson, Arizona, USA) with primary antibodies against the following proteins: cytokeratin 7 (CK7, 1:100; OV-TL12/30, Dako, Glostrup, Denmark), alpha-methylacyl-CoA racemase (AMACR, 1:100; 13H4, Dako), vimentin (1:200; V9, Dako), E-cadherin (1:25; NCH-38, Dako), epithelial membrane antigen (EMA, 1:500; GP1.4, Leica, Novocastra Laboratories, Newcastle upon Tyne, UK), CD10 (1:100; 56C6, Leica, Novocastra Laboratories), and Ki-67 (1:200; 30-9, Roche). The intensity of immunoreactivity was recorded as negative (0), weak (+), moderate (++), or strong (+++). The extent was recorded as the percentage of positive tumor cells. The Ki-67 index was assessed by the percentage of positive cells.
Statistical analysis
A χ2 test (Fisher’s exact test) or Student’s t-test was used to compare the differences between groups, and Kaplan–Meier curve analysis was used to compare survival between groups using SPSS Statistics 23.0 (IBM, Chicago, IL, USA).
Results
Clinicopathological features
The clinicopathological features are summarized in Table 1 and Supplementary Table 1. PRNRP was smaller than PRCC1 (p = 0.002) or PRCC2 (p < 0.001), respectively. All of the PRNRPs were staged as pT1a, whereas 65.2% of PRCC1s as pT1a (p = 0.006) and 61.5% of PRCC2s as pT1a (p = 0.003). None of the PRNRP cases showed renal vein thrombosis during surgery, microscopic lymphovascular invasion, renal capsular invasion, or pelvicalyceal system involvement. In addition, no PRNRP patients died of their PRNRP tumors during the follow-up period (Fig. 1).

Kaplan–Meier curve comparing disease-specific survival of patients with papillary renal neoplasm with reverse polarity (PRNRP) and patients with papillary renal cell carcinoma type 1 (PRCC1) and papillary renal cell carcinoma type 2 (PRCC2).
Targeted sequencing and PNA-mediated clamping PCR of KRAS
PRNRP tumors exhibited KRAS mutations in 25 out of the 26 cases (96%) by targeted sequencing (Fig. 2). The KRAS mutation types observed were G12V (16 cases), G12D (8 cases), and G12R (1 case). Other mutations found include MTOR (1 case, missense mutation), NF1 (1 case, nonsense mutation), POLE (1 case, frameshift insertion), ARID1A (1 case, missense mutation), and ARID1B (1 case, missense mutation) (Supplementary Table 1). When KRAS mutations were analyzed in 30 tumor and peritumoral non-tumor PRNRP tissues by PNA-mediated clamping PCR, mutations were observed in 28 out of the 30 (93%) PRNRP tumor samples. However, KRAS mutation was not found in all of the non-tumor tissues. The results of the KRAS mutation analysis by targeted sequencing of 26 PRNRPs agreed with those by PNA-mediated clamping PCR. None of the 23 PRCC1 and 26 PRCC2 samples revealed KRAS mutation in tumor nor non-tumor tissue.

Each column displays a patient; each row denotes a specific gene.
Comparative analysis with the TCGA dataset
In the TCGA dataset, five out of the 274 PRCC cases had a KRAS mutation. Three of them were found in PRCC2, and two in PRCC1. The mutation types observed from the TCGA dataset were similar to those observed in this study; three cases revealed G12D and two had G12V (Supplementary Table 2). Although there were some limitations in interpretation of the digital images of TCGA due to a limited number of slides to interpret and frozen section slides, three PRCC2 cases with KRAS mutations were quite similar to those of PRNRP. All of them showed a lower American Joint Committee on Cancer prognostic stage group (stage I) and pathologic tumor stage (pT1a or pT1). Two out of the 488 clear cell RCC cases exhibited KRAS mutations: one G12C, and the other A59G. Neither of the 81 chromophobe RCCs, nor the 62 unclassified RCCs revealed KRAS mutations [25, 26].
Microscopic features
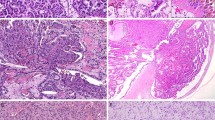
PRNRP cases showed lower Fuhrman nuclear and WHO/International Society of Urological Pathology (ISUP) grades than those observed in PRCC1 or PRCC2 cases (Table 2). Characteristically, the majority of the nuclei were located in the top or luminal area of the cells. Nuclear pseudostratification was noted less frequently in PRNRP than in PRCC2 (p < 0.001). In addition to these findings, the peritumoral lymphoid aggregation was more frequently noted in PRNRP than in PRCC1 (p < 0.001) and PRCC2 (p < 0.001) (Fig. 3). PRNRP revealed prominent papillary architecture (>80% of tumor). In the papillae, the hyalinized stroma was more frequently found in PRNRP than in PRCC1 (p < 0.001) and PRCC2 (p = 0.001). Edematous stroma, inflammatory cell infiltration, foamy macrophages in papillae, and old hemorrhages with degenerative change were observed to variable extents in PRNRP, PRCC1, and PRCC2. However, no PRNRP sample showed tumor necrosis. One PRNRP with wild KRAS (PRNRP_2, Supplementary Table 1) showed pseudostratification, mild nuclear size variation, and ISUP nucleolar grade 2. The immunohistochemical features were consistent with PRNRP, which were immunopositive for CK7, AMACR, and EMA. The other with wild KRAS (PRNRP_14, Supplementary Table 1) revealed typical histology with ISUP nucleolar grade 1, and typical immunohistochemical profile.

Papillary renal neoplasm with reverse polarity frequently exhibited a fibrous capsule with peritumoral lymphoid aggregation at lower power field (a). It revealed prominent papillary or tubulopapillary architecture (b), low-grade nuclei, inverted linear nuclear location (c, d), and hyalinized papillae (d). Papillary renal cell carcinoma type 1 disclosed single cell lining with low-grade nuclei (e) and papillary renal cell carcinoma type 2 showed abundant eosinophilic cytoplasm with high-grade nuclei (f).
Immunohistochemistry
Immunohistochemical profiles are summarized in Table 3. The intensity and extent of staining was homogenous in three cores of the same case. In PRNRP, most tumor cells were positive for CK7 and EMA, and variably positive for AMACR, E-cadherin and vimentin, whereas it was negative for CD10 (Fig. 4). Also, PRNRP exhibited lower Ki-67 labeling indices than PRCC1 and PRCC2.

The tumor cells were positive for cytokeratin 7 (a) and alpha-methylacyl-CoA racemase (b) and negative for CD10 (inset: matched normal renal tubules) (c). The Ki-67 immunostaining of papillary renal neoplasm with reverse polarity. Only a few cells are reactive for Ki-67. (d).
Discussion
The PRNRP is different from PRCC1 and PRCC2 in terms of molecular, clinicopathological, histological, and immunohistochemical features. The majority of PRNRPs were small, localized in the kidney, lower in pathologic stage, and showed indolent biologic behavior. Characteristic histological features of PRNRP were: papillary or tubulopapillary renal neoplasms with low-grade nuclei, inverted nuclear location, eosinophilic cytoplasm, hyalinized papillae, a single layer of epithelial cells (non-overlapped nuclei), no necrosis, and peritumoral lymphoid aggregation. PRNRP cases showed positive reactivity to CK7 and EMA and negative reactivity to CD10. The Ki-67 index of PRNRP was lower than that of PRCC1 and PRCC2.
In this study, we analyzed 90 cancer-related genes using targeted sequencing and found recurrent KRAS mutations in most of PRNRP cases, and verified the result by PNA-mediated clamping PCR. However, none of 23 PRCC1 and 26 PRCC2 exhibited a KRAS mutation. Recently, Al-Obaidy et al. reported recurrent KRAS mutations in eight of ten PRNRPs, and suggested it a characteristic finding of PRNRP [27]. In the TCGA dataset, PRCC reveals KRAS mutation in 5 out of 274 cases (1.8%), including three cases that are suggested to be PRNRP. The above findings suggest that recurrent KRAS mutation is a characteristic finding, if not an integral part of PRNRP.
PRNRP cases revealed G12V (16/26, 61.5%), G12D (9/26, 34.6%), and G12R (1/26, 3.9%) KRAS mutations. Somatic KRAS mutations are frequently found not only in adenocarcinomas of the lung, colon, and pancreas, but also in benign neoplasms, such as colorectal adenoma and sinonasal oncocytic papilloma [28,29,30,31]. The distribution of KRAS mutations differs according to the tumor type; G12C or G12V substitutions are frequently observed in lung adenocarcinoma, and G12D or G12V substitutions in both colorectal and pancreatic adenocarcinoma [32]. In turn, the downstream effects differ according to the substitution. The G12D substitution increases PI3K and MEK activation, while G12C or G12V induce RAS-related protein signaling and decrease AKT activation in non-small cell lung carcinoma cell lines [33].
Many genes are mutated in RCC cases. The frequently mutated genes of clear cell RCC are VHL (48.9%), PBRM1 (34.5%), SETD2 (12.9%), MUC4 (12.2%), BAP1 (10.1%), MUC16 (10.1%), MTOR (6.7%), and KDM5C (6.1%). The recurrently mutated genes of PRCC are TTN (15.2%), MUC16 (9.1%), MET (7.9%), KNT2C (6.6%), KIAA1109 (6.3%), SETD2 (5.9%), PKHD1 (5.7%), KMT2D (5.7%), OBSCN (5.2%), FAT1 (5.0%), and BAP1 (5.0%) [24, 25]. HLRCC-associated RCC is characterized by germline mutations in FH. MiT translocation RCC was characterized by Xp11 translocations involving either TFE3 or TFEB, and their partners are ASPSCR1, CLTC, DVL2, LUC7L3, KHSRP, MED15, RBM10, PRCC, PARP14, NONO, and SFPQ1 [34]. This shows that KRAS mutation is a unique finding of PRNRP, which is rarely observed in other type of RCCs.
Papillary renal tumors with oncocytic cytoplasm include PRNRP, PRCC2, HLRCC-associated RCC, MiT translocation RCC, acquired cystic disease-associated RCC, some morphologic variants of clear cell RCC, and rare collision tumors of oncocytic RCC [35]. Since PRNRP usually shows indolent biological behavior, it is important to differentiate PRNRP from other papillary tumors with oncocytic cytoplam. Differentiation of PRNRP from PRCC2 is sometimes difficult, because both of them may reveal similar histologic findings. In cases with overlapping histology of PRCC2 and PRNRP, precise histomorphologic examination, immunohistochemical staining, and KRAS mutation study may help for making a correct diagnosis. In addition, immunoreactivity for GATA3 and L1CAM could be helpful for identifying PRNRP [13]. Although reverse polarity of nuclei is one of the characteristic features of PRNRP, it is found in PRCC2 or MiT translocation RCC. In this study, even though more than 20% of the PRCC2 cases show focally inverted nuclei, we could identify them depending on other nuclear findings (such as high-grade nuclear features, or pseudostratification) and immunohistochemical findings. Since subsets of RCCs with MiT aberrations may reveal oncotyic and papillary RCC like morphology, it is necessary to exclude those cases by FISH assays [14].
Since the introduction of the two groups of PRCCs as PRCC1 and PRCC2, several studies have examined the cases that do not meet the criteria of the two groups [3, 4, 36, 37]. Some investigators have named those cases “PRCC, not otherwise specified”, “type 3 PRCC”, or “unclassified PRCC”. Recently, next-generation sequencing-based studies showed that PRCC is composed of at least four groups that have different clinical and molecular profiles [4, 5]. PRCC2 is divided into three molecular subgroups with different prognoses [4]. Because both PRNRP and PRCC2 contain cells with abundant and eosinophilic cytoplasms, it is sometimes difficult to differentiate them. In a whole-genome sequencing PRCC study, PRNRP was included in PRCC and was stratified as “C2a”, a PRCC2 cluster with early-stage and good prognosis [4].
This study has several limitations. Since this study was performed using a ready-made cancer-related gene panel for targeted sequencing, some of the renal cancer-related genes (such as FH, BAP1, SDH, etc.) were not included. Secondly, TFE3 and TFEB FISH analysis was not performed to exclude RCC with MiT aberrations. Thirdly, the quality and quantity of TCGA images that we observed were not good. We interpreted only a limited number of images (two or three images per case) and some of them were images of frozen section slides. Finally, the function of mutated KRAS in the pathogenesis or progression of PRNRP was not studied.
In conclusion, PRNRP is a subtype of PRCC characterized by KRAS mutation, oncocytic cytoplasm, low-grade nuclear features, inverted nuclear location, and indolent biologic behavior. These findings could be used as a key to identify PRNRP, especially in cases with overlapping histology between PRCC2 and PRNRP.
References
Amin MB, Amin MB, Tamboli P, Javidan J, Stricker H, de-Peralta Venturina M, et al. Prognostic impact of histologic subtyping of adult renal epithelial neoplasms: an experience of 405 cases. Am J Surg Pathol. 2002;26:281–91.
Delahunt B, Eble JN. Papillary renal cell carcinoma: a clinicopathologic and immunohistochemical study of 105 tumors. Mod Pathol. 1997;10:537–44.
Saleeb RM, Brimo F, Farag M, Rompre-Brodeur A, Rotondo F, Beharry V, et al. Toward biological subtyping of papillary renal cell carcinoma with clinical implications through histologic, immunohistochemical, and molecular analysis. Am J Surg Pathol. 2017;41:1618–29.
Cancer Genome Atlas Research N, Linehan WM, Spellman PT, Ricketts CJ, Creighton CJ, Fei SS, et al. Comprehensive molecular characterization of papillary renal-cell carcinoma. N Engl J Med. 2016;374:135–45.
Ricketts CJ, De Cubas AA, Fan H, Smith CC, Lang M, Reznik E, et al. The cancer genome atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 2018;23:313–26 e5.
Delahunt B, Algaba F, Eble J, Cheville J, Amin MB, Argani P, et al. Papillary renal cell carcinoma. In: Moch H, Humphrey PA, Ulbright TM, Reuter VE, editors. WHO classifications of tumours of the urinary system and male genital organs. 4th ed. Lyon: International Agency for Research on Cancer; 2016. p. 23–4.
Lefevre M, Couturier J, Sibony M, Bazille C, Boyer K, Callard P, et al. Adult papillary renal tumor with oncocytic cells: clinicopathologic, immunohistochemical, and cytogenetic features of 10 cases. Am J Surg Pathol. 2005;29:1576–81.
Kunju LP, Wojno K, Wolf JS Jr., Cheng L, Shah RB. Papillary renal cell carcinoma with oncocytic cells and nonoverlapping low grade nuclei: expanding the morphologic spectrum with emphasis on clinicopathologic, immunohistochemical and molecular features. Hum Pathol. 2008;39:96–101.
Xia QY, Rao Q, Shen Q, Shi SS, Li L, Liu B, et al. Oncocytic papillary renal cell carcinoma: a clinicopathological study emphasizing distinct morphology, extended immunohistochemical profile and cytogenetic features. Int J Clin Exp Pathol. 2013;6:1392–9.
Han G, Yu W, Chu J, Liu Y, Jiang Y, Li Y, et al. Oncocytic papillary renal cell carcinoma: a clinicopathological and genetic analysis and indolent clinical course in 14 cases. Pathol Res Pr. 2017;213:1–6.
Hes O, Brunelli M, Michal M, Cossu Rocca P, Hora M, Chilosi M, et al. Oncocytic papillary renal cell carcinoma: a clinicopathologic, immunohistochemical, ultrastructural, and interphase cytogenetic study of 12 cases. Ann Diagn Pathol. 2006;10:133–9.
Park BH, Ro JY, Park WS, Jee KJ, Kim K, Gong G, et al. Oncocytic papillary renal cell carcinoma with inverted nuclear pattern: distinct subtype with an indolent clinical course. Pathol Int. 2009;59:137–46.
Al-Obaidy KI, Eble JN, Cheng L, Williamson SR, Sakr WA, Gupta N, et al. Papillary Renal neoplasm with reverse polarity: a morphologic, immunohistochemical, and molecular study. Am J Surg Pathol. 2019;43:1099–111.
Skala SL, Xiao H, Udager AM, Dhanasekaran SM, Shukla S, Zhang Y, et al. Detection of 6 TFEB-amplified renal cell carcinomas and 25 renal cell carcinomas with MITF translocations: systematic morphologic analysis of 85 cases evaluated by clinical TFE3 and TFEB FISH assays. Mod Pathol. 2018;31:179–97.
Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–60.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Lai Z, Markovets A, Ahdesmaki M, Chapman B, Hofmann O, McEwen R, et al. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016;44:e108.
Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–9.
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–11.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6:80–92.
Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6.
Skidmore ZL, Wagner AH, Lesurf R, Campbell KM, Kunisaki J, Griffith OL, et al. GenVisR: genomic visualizations in R. Bioinformatics. 2016;32:3012–4.
Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43:D805–11.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Disco. 2012;2:401–4.
Al-Obaidy KI, Eble JN, Nassiri M, Cheng L, Eldomery MK, Williamson SR, et al. Recurrent KRAS mutations in papillary renal neoplasm with reverse polarity. Mod Pathol. 2019. https://www.nature.com/articles/s41379-019-0362-1. Accessed 22 Oct 2019.
Eberhard DA, Johnson BE, Amler LC, Goddard AD, Heldens SL, Herbst RS, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol. 2005;23:5900–9.
Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–34.
Wiland HOt, Shadrach B, Allende D, Carver P, Goldblum JR, Liu X, et al. Morphologic and molecular characterization of traditional serrated adenomas of the distal colon and rectum. Am J Surg Pathol. 2014;38:1290–7.
Udager AM, McHugh JB, Betz BL, Montone KT, Livolsi VA, Seethala RR, et al. Activating KRAS mutations are characteristic of oncocytic sinonasal papilloma and associated sinonasal squamous cell carcinoma. J Pathol. 2016;239:394–8.
Ostrow SL, Simon E, Prinz E, Bick T, Shentzer T, Nagawkar SS, et al. Variation in KRAS driver substitution distributions between tumor types is determined by both mutation and natural selection. Sci Rep. 2016;6:21927.
Ihle NT, Byers LA, Kim ES, Saintigny P, Lee JJ, Blumenschein GR, et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst. 2012;104:228–39.
Pei J, Cooper H, Flieder DB, Talarchek JN, Al-Saleem T, Uzzo RG, et al. NEAT1-TFE3 and KAT6A-TFE3 renal cell carcinomas, new members of MiT family translocation renal cell carcinoma. Mod Pathol. 2019;32:710–6.
Kryvenko ON, Jorda M, Argani P, Epstein JI. Diagnostic approach to eosinophilic renal neoplasms. Arch Pathol Lab Med. 2014;138:1531–41.
Yang XJ, Tan MH, Kim HL, Ditlev JA, Betten MW, Png CE, et al. A molecular classification of papillary renal cell carcinoma. Cancer Res. 2005;65:5628–37.
Alomari AK, Nettey OS, Singh D, Kluger H, Adeniran AJ. Clinicopathological and immunohistochemical characteristics of papillary renal cell carcinoma with emphasis on subtyping. Hum Pathol. 2015;46:1418–26.
Acknowledgements
The biospecimens and data used for this study were provide by the Biobank of Chonnam National University Hwasun Hospital, a member of the Korea Biobank Network.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Kim, S.S., Cho, Y.M., Kim, G.H. et al. Recurrent KRAS mutations identified in papillary renal neoplasm with reverse polarity—a comparative study with papillary renal cell carcinoma. Mod Pathol 33, 690–699 (2020). https://doi.org/10.1038/s41379-019-0420-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-019-0420-8
This article is cited by
-
Genomic alterations and diagnosis of renal cancer
Virchows Archiv (2024)
-
Ipsilateral synchronous papillary renal neoplasm with reverse polarity and urothelial carcinoma in a renal transplant recipient: a rare case report with molecular analysis and literature review
Diagnostic Pathology (2023)
-
Papillary renal neoplasm with reverse polarity may be a novel renal cell tumor entity with low malignant potential
Diagnostic Pathology (2022)
-
Recurrent KRAS mutations are early events in the development of papillary renal neoplasm with reverse polarity
Modern Pathology (2022)
-
Classification of renal cell tumors – current concepts and use of ancillary tests: recommendations of the Brazilian Society of Pathology
Surgical and Experimental Pathology (2021)
