
Abstract
Sinonasal mucosal melanoma is a rare tumor arising within the nasal cavity, paranasal sinuses, or nasopharynx (sinonasal tract). This study evaluated 90 cases diagnosed in 29 males and 61 females with median age 68 years. Most tumors involved the nasal cavity and had an epithelioid morphology. Spectrum of research techniques used in this analysis includes targeted-DNA and -RNA next-generation sequencing, Sanger sequencing, fluorescence in situ hybridization and immunohistochemistry. Sinonasal melanomas were commonly driven by RAS (38/90, 42%), especially NRAS (n = 36) mutations and rarely (4/90, 4%) displayed BRAF pathogenic variants. BRAF/RAS mutants were more frequent among paranasal sinuses (10/14, 71%) than nasal (26/64, 41%) tumors. BRAF/RAS-wild type tumors occasionally harbored alterations of the key components and regulators of Ras-MAPK signaling pathway: NF1 mutations (1/17, 6%) or NF1 locus deletions (1/25, 4%), SPRED1 (3/25, 12%), PIK3CA (3/50, 6%), PTEN (4/50, 8%) and mTOR (1/50, 2%) mutations. These mutations often occurred in a mutually exclusive manner. In several tumors some of which were NRAS mutants, TP53 was deleted (6/48, 13%) and/or mutated (5/90, 6%). Variable nuclear accumulation of TP53, mirrored by elevated nuclear MDM2 expression was seen in >50% of cases. Furthermore, sinonasal melanomas (n = 7) including RAS/BRAF-wild type tumors (n = 5) harbored alterations of the key components and regulators of canonical WNT-pathway: APC (4/90, 4%), CTNNB1 (3/90, 3%) and AMER1 (1/90, 1%). Both, TERT promoter mutations (5/53, 9%) and fusions (2/40, 5%) were identified. The latter occurred in BRAF/RAS-wild type tumors. No oncogenic fusion gene transcripts previously reported in cutaneous melanomas were detected. Eight tumors including 7 BRAF/RAS-wild type cases expressed ADCK4::NUMBL cis-fusion transcripts. In summary, this study documented mutational activation of NRAS and other key components and regulators of Ras-MAPK signaling pathway such as SPRED1 in a majority of sinonasal melanomas.
Similar content being viewed by others

Introduction
Sinonasal tract mucosal melanoma (SNTMM), first reported in 1869, consist of tumors developing in the nasal cavity, paranasal sinuses, and nasopharynx1,2. SNTMM is an aggressive tumor mostly diagnosed in sixth and seventh decades of life. In the United States, it accounts for <1% of all melanomas, with a steady increased incidence in white females3.
Histologically, SNTMM has an epithelioid, spindle cell, or round cell/undifferentiated morphology. A lack of melanin pigmentation is common. Therefore, immunohistochemical demonstration of melanocytic markers is essential for the diagnosis. However, the immunohistochemical profile of SNTMMs and cutaneous tumors is similar and cannot distinguish primary lesion from metastatic ones2.
SNTMM is often diagnosed at an advanced stage, with surgery as the first-line treatment, but only 25–30% of patients survive more than five-years. As a result, adjuvant therapy, including postoperative radiation, chemotherapy and targeted therapy are often considered4. As more options become available for the latter, the need to identify genetic markers has become increasingly important.
The mutation profile of SNTMM remains incompletely characterized. Most Sanger sequencing studies have been limited to BRAF, KIT, and NRAS mutation status5,6,7,8,9,10,11. Recent investigations utilizing targeted-, whole exome-, and whole genome-next generation sequencing (NGS) provided more comprehensive mutation profiles but with only a limited number of cases analyzed12,13,14,15,16,17.
The aim of this study was to elucidate the molecular characteristics of SNTMM. A large series of well-characterized tumors was evaluated using targeted NGS, Sanger sequencing, fluorescence in-situ hybridization (FISH), and immunohistochemistry (IHC), identifying genetic alterations affecting dominant oncogenes and tumor suppressor genes and their respective pathways in SNTMM.
Material and methods
Following review of clinical data, evaluation of histopathology and immunohistochemistry, and assessment of the quality of nucleic acids, 90 cases (80 primary tumors and 10 local recurrences) were included in the study. A process of tumor selection is described in detail in Supplementary Data.
Immunohistochemical studies
Immunohistochemistry (IHC) was performed using either BenchMark Ultra (Ventana Medical Systems-Roche Group, Tucson, AZ) or Leica Bond-Max automated immunostainer (Leica, Bannockburn, IL). A threshold of ≤10% was used for focal positivity, while >10% but ≤80% and >80% for mosaic and diffuse positivity, respectively. The panel included antibodies against human melanoma markers, PReferentially expressed Antigen in MElanoma (PRAME), keratin proteins, histone H3 trimethylated at lysine 27 (H3K27me3), transcription factor E3 (TFE3), synaptophysin (SYP), DNA-mismatch repair (MMR) proteins, β-catenin (CTNNB1), C-myc (CMYC), KIT (CD117), mouse double minute 2 (MDM2), tumor protein 53 (TP53), neurotrophic tyrosine receptor kinase (NTRK) and ROS proto-oncogene 1 (ROS-1). Detailed description of antibodies and protocols is provided in Supplemental Material and Methods and Supplementary Table 1.
Genetic studies
Nucleic acids were extracted from formalin fixed paraffin embedded (FFPE) tumor tissues using Maxwell® RSC system (Promega, Madison, WI). Targeted-DNA NGS was done employing Ion Torrent™ (Life Technologies/Thermo Fisher Scientific, Waltham, MA) platform and either cancer hotspot (CH) or comprehensive cancer (CC) gene panels. Sprouty Related EVH1 Domain Containing 1 (SPRED1) was evaluated using custom made Ion AmpliSeq™ libraries. Targeted-RNA NGS for the detection of fusion gene transcripts were done using Archer® FusionPlex Solid Tumor panel (ArcherDx, Boulder, CO) and MiSeqDx sequencing instrument (Illumina, San Diego, CA). Genes targeted by NGS are listed in Supplementary Table 2. Telomerase reverse transcriptase gene promoter (TERTp) region frequently harboring mutations (228 C > T and 250 C > T) was screened by conventional PCR amplification and Sanger sequencing. Integrity of NF1 and TP53 loci were evaluated by interphase FISH using TP53/NF1 deletion probe (MetaSystems Probes GmbH, Altlussheim, Germany). Detailed NGS, Sanger sequencing, and FISH protocols are available in Supplemental Material and Methods.
Results
Demographic and clinicopathologic data
SNTMMs analyzed in this study were diagnosed in Europe (n = 67), USA (n = 18) and Japan (n = 5). There were 29 males and 61 females (ratio 1:2.1). The median age at the diagnosis was 68 years (67 years for females and 70 years for males). Nine of 73 tumors localized within the nasal cavity also expanded into the maxillary (n = 6) or ethmoid sinus. Three melanomas involved the nasopharynx. In 13 cases, tumors affected a single paranasal sinus: maxillary (n = 9), ethmoid (n = 3), and frontal sinus; one tumor affected both maxillary and ethmoid sinuses. None of the patients had a history of primary melanoma elsewhere in the body. Demographic and clinicopathologic data for each patient are listed in Supplementary Table 3.
Histological features and immunohistochemical profile
Exclusively epithelioid morphology (n = 57) or predominantly epithelioid morphology with focal spindle (n = 12) or round cell (n = 9) pattern was seen in 87% of SNTMMs. One tumor had all three patterns. Remaining 11 cases displayed spindle cell or mostly spindle cell features. Nuclear pleomorphism was present in 59% (53/90) and necrosis in 48% (43/90) of cases. Mitoses (evaluated in 87 cases with sufficient tumor volume) varied from 2 to 132 (median 20) per 2 mm2. Melanin pigmentation was seen in 58% (52/90) of tumors, focally in 10 cases. Histopathologic features analyzed for each case are listed in Supplementary Table 3. Representative histopathological images are shown in Fig. 1A–C.

A Tumor composed of uniform rounded cells with prominent nucleoli. B A spindle cell variant with scant collagenous matrix. C An epithelioid tumor with pigmentation. D Prominent nuclear expression of SOX10 seen in 99% (88/89) of cases.
Expression of antigens was assessed using IHC. All SNTMMs were positive for at least one melanocytic marker (HMB-45, MELAN A, S100 protein, SOX10, Tyrosinase). A representative immunohistochemical image is shown in Fig. 1D. Synaptophysin was detected in 26% (20/78) of SNTMMs with 12 showing immunoreactivity in more than 10% of cells. Three cases (4%) revealed focal keratin expression. At least partial nuclear retention of H3K27me3 was seen in all analyzed (n = 82) cases, with a significant fraction (33/82, 40%) demonstrating mosaic staining. Almost all (78/79, 99%) SNTMMs showed PRAME-immunopositivity with diffuse expression pattern seen in 89% (69/79) of cases. None of analyzed (n = 79) tumors expressed TFE3. Detailed results of immunohistochemical studies are listed in Supplementary Table 4.
Overview of targeted-DNA NGS
Molecular status of 50 oncogenes and tumor suppressor genes was assessed in all SNTMMs. Additional sequencing data of 359 genes were available for 21 cases. SPRED1 was sequenced separately in 50 SNTMMs including 25 BRAF/RAS-WT tumors. Of 156 detected sequence variations, 124 represented unique molecular events including single nucleotide substitutions (n = 102), deletions or deletion-insertions (n = 17), two duplications (n = 2), and insertions (n = 3). Some of these alterations triggered frameshift (n = 9) or STOP-codon (n = 12) mutations.
Almost half (n = 60) of sequence variations were previously identified to occur in somatic manner in cancer. Of remaining alterations 30% (19/64) had a variant allelic frequency (VAF) 40–60% that might indicate germline nature18. Nevertheless, normal tissue matching tumor samples were not available and a direct assessment of somatic versus germline status was not possible.
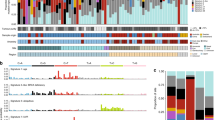
Most important molecular findings including gene mutations/sequence variations detected by NGS are summarized in Fig. 2. Supplemental Database shows all mutations/sequence variations (sheet A) and their pathogenic effects (sheet B) calculated/predicted by FATHMM, PolyPhen, SIFT and Human Genomic Variant Search Engine (https://varsome.com).

Clinical data abbreviations: F female, M male, N nasal cavity, NS nasal cavity with sinus involvement, S sinus, NP nasopharynx, P primary, R recurrence. Results of molecular and FISH studies are marked by colors. Green indicates mutations/sequence variations detected by NGS: cancer hot spot panel (CH)-grass green, comprehensive cancer panel (CC)-spruce green and SPRED1 panel-pastel green, empty box-missense mutation, d -in frame deletion, f*-STOP codon/frameshift mutation, n-NRAS mutation, k-KRAS, l-low allele frequency. Blue box indicates NF1/TP53 FISH data. Yellow indicates changes detected by ArcherDx: cis-cis fusion, i-oncogenic isoform, t-TERT fusion, nf-likely not in frame). Brown box indicates TERT Sanger sequencing data (u- unsuccessful PCR amplification but preserved DNA template). Black box indicates alterations detected by FISH and NGS. Other: 2 = two mutations, nd not done.
BRAF, RAS, NF1 and SPRED1
BRAF variants were found in 4% (4/90) of SNTMMs. They included class 2 (p.K601N, p.G469A) and class 3 (p.S467L) mutations, and p.V471I substitution of unknown significance. The last two coincided with NRAS p.G12A driver mutations and showed low (≤10%) allele frequency. No p.V600E class 1 mutation (typical of cutaneous melanoma) was detected. However, transcripts of BRAF isoform containing exon 10 to 18 duplication were identified in the tumor harboring GNA11 p.Q209L mutation.
RAS driver mutations were identified in 42% (38/90) of SNTMMs. NRAS (n = 36) mutations involved codon 61 (n = 16), 12 (n = 14) and 13 (n = 6) with p.Q61K substitution being the most common (n = 8). NRAS mutants carried additional mutations/sequence variations in 39% (14/36). Affected genes encoded receptor tyrosine kinases (RTKs) other than KIT, G-protein α subunits, components of the PI3K-, RB1-, TP53-, WNT- pathways, chromatin regulatory factors and proteins participating in DNA repair processes. Two SNTMMs harbored KRAS hot-spot mutations, p.G12D and p.G13D. The former coincided with JAK2 p.G614E substitution. BRAF/RAS mutations were identified in 71% (10/14) of primary paranasal sinuses melanomas but only in 41% (26/64) of nasal cavity tumors. Also, no such mutations were seen in three lesions involving nasopharynx.
NF1 mutations (n = 3) were detected in 10% (2/21) of SNTMMs. A NRAS mutant revealed two NF1 truncating (p.Q1070*, p.S2188fs) mutations, while a BRAF/RAS-WT tumor harbored NF1 p.G1219R substitution coinciding with mutations/sequence variations in CDKN2A, EGFR, MTOR, TERTp, and TP53.
SPRED1 frameshift mutations (p.E51*, p.Q158Sfs*15, p.V309Wfs*9) were identified in 12% (3/25) of BRAF/RAS-WT SNTMMs including KIT mutant. In contrast, 1 of 25 BRAF/RAS-mutants harbored low allele frequency SPRED1 variant (p.R207K).
BRAF/RAS/NF1-WT
Mutations/sequence variations in genes encoding RTKs, G-protein α subunits, components of the PI3K-, RB1-, TP53-, WNT- pathway (PIK3CA p. E542K, RB1 p.V754L, TP53 p.G244D, CTNNB1 p.S45del), chromatin regulatory factors (EZH2 p.Y646C) and proteins participating in DNA repair processes (ERCC4 p.R799W) were found in 36% (12/33) of BRAF/RAS-WT (evaluated with Ion Torrent™ CH-panel) and 88% (14 of 16) of BRAF/RAS/NF1-WT tumors (Ion Torrent™ CC-panel). A complete list of identified genetic changes is available in Supplemental Database.
GNAS, GNAQ and GNA11
Mutations in GNAS, GNAQ and GNA11, genes encoding G-protein α subunits, were identified in 6% (5/90) of SNTMMs. However, only GNA11 p.Q209L demonstrated high allele frequency (58%). GNAQ (n = 4) and GNAS (n = 2) mutations identified in 4 melanomas showed low allele frequency and co-occurred with mutations affecting PI3K pathway.
PI3K-AKT-MTOR
Mutations/sequence variations in genes encoding key components of PI3K pathway, AKT1, PIK3CA (n = 5) and PTEN (n = 4) were identified in 11% (10/90) of SNTMMs. Additional sequencing data obtained in 21 cases evaluated with the Ion AmpliSeq™ CC-panel revealed mutations in MTOR, PIK3C2B (n = 2), PIK3CG, PIK3R2 and TSC2. The latter coincided with PIK3C2B mutation. Three of 14 mutations affecting PI3K pathway occurred in NRAS mutants.
KIT
Gain-of-function KIT p.K642E mutation was detected in 2% (2/90) of SNTMMs. In both cases, the KIT mutation coincided with mutation activating other oncogene (CTNNB1) or inactivating tumor suppressor gene (SPRED1).
Receptor tyrosine kinases other than KIT
Fifteen sequence variations affecting CSF1R, EGFR (n = 3), ERBB2, FGFR3 (n = 2), KDR, MET (n = 3), NTRK3, RET, ROS-1 and TSHR, genes encoding RTKs, were identified in 13 SNTMMs. A protein kinase domain was frequently (n = 10) involved. Computational techniques predicted damaging potential of all substitutions. RTK sequence variations coincided with PI3K (n = 5), and TP53 (n = 4) pathway alterations and NRAS hot-spot mutations (n = 4).
Cell cycle related genes, CDKN2A, TP53 and RB1
TP53 mutations were identified in 6% (5/90) of SNTMMs. TP53 alterations coincided with NRAS drivers (n = 4) and mutations affecting genes encoding RTKs (n = 5), canonical WNT pathway (n = 2), RB1, and CDKN2A. In one tumor evaluated with the Ion AmpliSeq™ CH-panel, TP53 p.G244D substitution was the sole genetic abnormality.
Canonical WNT/Beta-catenin pathway
Mutations in CTNNB1 (n = 3) and APC (n = 4), genes encoding components of canonical WNT pathway, were detected in 7% (6/90) of tumors. In three tumors, APC and/or CTNNB1 mutation coincided with NRAS (n = 2) or KIT driver mutations. One of 21 SNTMMs evaluated with the Ion AmpliSeq™ CC-panel revealed mutation in AMER1 (APC Membrane Recruitment Protein 1), a canonical WNT pathway regulator. This Triple-WT tumor also harbored mutations/sequence variations in several epigenetic modifiers and components of PI3K pathway and CNOT4::TERT fusion transcripts.
Epigenetic modifiers and DNA repair pathways
SNTMMs harbored mutations/sequence variations in various genes involved in epigenetic processes such as chromatin structure regulation, transcriptional repression, or DNA repair processes. Among mutated genes encoding chromatin regulatory factors (CRF) were SMARCB1 encoding component of ATP-dependent chromatin remodeling complex, EZH2 and KMT2C encoding histone tail modifiers, TET2 encoding DNA demethylase, IDH1 encoding an enzyme that generates metabolite inhibiting CRFs and BRD3 encoding protein associated with histone acetylation. All mutations except for BRD3 p.K655R substitution represented unique molecular events. CRF mutants often lacked mutations in canonical melanoma drivers.
Mutations/sequence variations affecting various DNA repair pathways were identified in 10% (9/90) of SNTMMs including five BRAF/RAS/NF1-wild type tumors and four NRAS mutants. FANCA, FANCC, and ERCC4, components of the Fanconi anemia (FA) damage repair pathways were mutated in three cases. Two SNTMMs including a ERCC4 mutant, harbored an identical frameshift mutation in XRCC2, a RAD51 paralog involved in homologous recombination repair of DNA damage. Mutations affecting members of DNA damage response MRE11-RAD50-NBS1 complex were detected in 2 other tumors. ATR, MUTYH, RECQL4 and PMS1, which function in various DNA repair pathways were mutated in four cases, one mutation in each tumor.
TERTp Sanger sequencing study
Two TERTp mutation hot spots were evaluated in 68 tumors. Of successfully PCR amplified samples, 9% (5/54) revealed c.228 C > T (n = 4) and c.250 C > T TERTp mutations. PCR failed to produce amplification products in 21% (14/68) of analyzed cases, although DNA/RNA quality controls indicated well-preserved nucleic acids.
Fusion gene transcripts
Forty SNTMMs were evaluated for fusion gene transcripts. Two tumors (5%) revealed TERT fusion with either CNOT4 or NUP50. DCAF7(e1)::PRKCA(e3) fusion transcripts, predicted to be not in-frame, were detected in one case. Eight (20%, 8/40) SNTMMs including 7 BRAF/RAS-WT tumors expressed ADCK4(e15)::NUMBL(e3) cis-fusion transcripts.
NF1 and TP53 loci FISH study
Integrity of NF1 and TP53 loci were evaluated by FISH in 48 cases. Deletion of NF1 locus was a sole molecular change in one tumor. Heterozygous TP53 locus deletion was detected in 13% (6/48) of tumors.
Immunohistochemical analysis of pathways and oncogenes
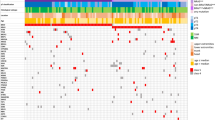
CMYC was commonly expressed, but 13% (11/82) of tumors were negative. KIT expression varied from focal to diffuse and was detected in 56% (46/82) of cases. Both KIT mutants revealed diffuse, strong KIT immunoreactivity (Fig. 3A). Most SNTMMs had prominent membranous and weak cytoplasmic β-catenin staining. However, four tumors, including three CTNNB1 mutants, showed strong cytoplasmic and focal or diffuse nuclear staining (Fig. 3B). Nuclear TP53 immunoreactivity was seen in 87% (71/82) of SNTMMs. The most common reaction was mosaic staining (>10 to ≤80% of positive nuclei) detected in 34% of cases. Nineteen (23%) SNTMMs had diffuse (>80%) TP53 expression, while focal (≤10% of positive nuclei) immunoreactivity was seen in 24 (29%) cases. No TP53 staining was detected in 11 SNTMMs including two cases harboring TP53 alterations. Diffuse or mosaic MDM2 nuclear staining was seen in 40% (33/82) tumors. The pattern of MDM2 expression mirrored TP53 staining in most cases (Fig. 3C, D). Furthermore, NTRK and ROS1 expression was evaluated in tumors harboring NTRK3 and ROS1 mutations, respectively. NTRK3 mutant revealed focal NTRK expression, while ROS1 mutant lacked ROS1 immunoreactivity.

A Membrane and dot-like cytoplasmic KIT(CD117) expression in p.K642E mutant. B Beta-catenin nuclear accumulation in tumors harboring CTNNB1 p.S45del. C Nuclear accumulation of TP53, mirrored by elevated nuclear expression of MDM2 (D) in NRAS p.G12D mutant.
Mismatch repair (MMR) proteins were evaluated in 78 SNTMMs. The loss of MLH1 and PMS2 expression was documented in three BRAF/RAS-WT SNTMMs. These tumors harbored, respectively, EZH2, PTEN, and TP53 mutation/sequence variation. MSH2 and MSH6 expression was retained in all cases. None of SNTMMs harbored MLH1 mutation. Immunohistochemistry results (CTNNB1, CMYC, KIT, MDM2, TP53 and MMR proteins) for each case are available in Supplementary Table 5.
Discussion
Mucosal melanoma is a rare melanoma subtype. In general, mucosal tumors are characterized by low point mutation burden and high number of structural variants. Recent studies indicated that tumors arising in different organs may have different mutation profiles15,16,17.
This study evaluated 90 well-documented SNTMMs using spectrum of molecular techniques including targeted-NGS, Sanger sequencing and FISH. Unfortunately, copy number variation analysis was not available for this investigation. All cases with a clinical history of a primary cutaneous or another mucosal melanoma were excluded, considering that the SNT is an uncommon site of melanoma metastases19. Primary SNT malignant peripheral nerve sheath tumor and perivascular epithelioid cell tumor was considered in the differential diagnosis and excluded based on morphology and immunophenotype, including retention of H3K27me3 or lack of nuclear TFE3 expression20,21.
The BRAF/MEK/ERK signaling pathway, known as the mitogen-activated-protein-kinase (MAPK) pathway and PI3K/AKT/mTOR pathway promotes cell proliferation and survival22. Gain-of-function BRAF or RAS mutations and NF1 inactivation account for pathologic signaling of these pathways23. Recently developed cutaneous melanoma molecular classification specified four subtypes: BRAF-, RAS-, NF1-mutants, and Triple-WT tumors24. The latter is defined as group of molecularly heterogenous tumors which lacks BRAF and RAS hot-spot mutations and NF1 inactivation.
BRAF p.V600E substitution, a hallmark mutation of cutaneous melanoma was not detected in this cohort. However, one tumor harbored mutation in p.K601, another BRAF hot-spot24. In SNTMM, hot-spot and non-hot-spot BRAF mutations are occasionally reported, the latter co-occurring with canonical RAS mutations14,17.
NRAS mutations were found in 42% of SNTMM, predominantly paranasal tumors. Previous studies of 10 or more SNTMMs reported NRAS mutant frequency in the range of 7–22% and 26–54% for Sanger and NGS, respectively5,6,8,9,10,11,12,13,14,17,25. Also, the involvement of codons 61, 12, and 13 mirrored published Sanger sequencing and NGS data5,6,8,10,13,14,17,25,26,27,28. However, a very recent investigation reported 50% (8/16) of NRAS mutations in codons 7, 8, 17, 58, 62, 63, and 659. Similar mutations have been described in a few tumors, including cutaneous melanomas but not in sinonasal or other mucosal melanomas. Detailed analysis is presented in Supplementary Table 6. KRAS hot-spot mutations are rare in mucosal melanoma15,16,17,29. In SNTMMs, only two cases in combined 44 SNTMMs were reported11,14. Similarly, only two such mutants were found in this cohort.
NF1, encoding a negative regulator of RAS, is the third most frequent mutated gene in UV-signature melanomas after BRAF and NRAS30. NF1 mutations were identified in 25 to 31% of SNTMMs in relatively small cohorts of tumors by whole exome- and whole genome- sequencing studies16,17. This investigation utilizing targeted NGS, found NF1 mutations in 10% of cases, similar to a previous SNTMM study employing a similar sequencing strategy14. Early studies suggested that NF1 alterations were mutually exclusive with BRAF and RAS mutations30. However, the co-occurrence of NF1 and non-hot-spot BRAF and hot-spot RAS mutations has been documented24. The latter was seen in this study and reported in head and neck mucosal melanomas including SNT tumors29,31. NF1-mutant cutaneous melanoma was associated with male sex and older age at diagnosis24,32. A similar correlation is not seen in SNTMM harboring NF1 alterations. Clinicopathologic characteristics of 13 NF1 mutant SNTMMs identified by the current and previous studies is presented in Table 1.
SPRED1, member of SPRED protein family, acts as another negative regulator of Ras-MAPK signaling binding directly to c-KIT and RasGAP. Loss-of-function mutations in SPRED1 leads to the developmental disorder, Legius syndrome, and were reported in human cancer33. More recent studies documented SPRED1 inactivation in mucosal melanomas predominantly in anorectal (67%) and vulvovaginal (33%) and less frequently in sinonasal tumors (12.5%)12. In this cohort of SNTMMs, SPRED1 frameshift/stop codon mutations were identified in 12% (3/25) of BRAF/RAS-WT tumors and their occurrence were mutually exclusive to mutations affecting components of Ras-MAPK signaling pathway such as PIK3CA and PTEN as previously reported12. However, SPRED1 mutation coincided with activating KIT mutation in 1 of 3 cases. The latter was seen in anorectal and vulvovaginal tumor but not in SNTMM12.
Gain of function KIT mutations or gene amplification were reported in mucosal and acral melanomas and those arising in chronically UV-exposed skin. Most of KIT oncogenic mutations were detected in exon 11 and 13 with p.L576P and p.K642E substitutions being quite common34,35. In this study, 2% (2/90) of SNTMMs harbored KIT p.K642E mutation. The frequency of KIT mutations in SNTMMs appears to be low (5%; 13/246) based on the current and published studies of 10 or more cases5,6,8,10,11,13,14,17,25. A very recent investigation reported KIT mutations in 22% (16/72) of SNTMMs. Furthermore, exon 11 deletions (n = 4) and duplication accounted for 31% (5/16) of these mutations9. Such alterations are exceedingly rare in mucosal melanomas and represent <10% of all KIT mutations6,25,29,36,37,38,39,40,41,42,43,44. Frequency of reported types of KIT mutations in mucosal melanomas is presented in Supplementary Table 7. KIT expression was documented by IHC in mucosal melanomas including sinonasal tumors5,9,39. In this cohort, KIT immunoreactivity was common and did not correlate with mutation status. Also, there was no association between KIT and CMYC positivity and pigmentation as previously reported9. However, KIT mutants revealed high mitotic rates. Previous evaluation reported a correlation between KIT mutations and increased cell proliferation rate in metastatic oral melanomas45.
This study documented the dominant role of NRAS versus KIT oncogenesis in SNTMMs. This observation was corroborated by the review of RAS and KIT mutations reported in head and neck melanomas5,6,8,9,10,13,14,16,17,25,27,28. However, a reverse correlation between RAS and KIT mutations has been reported in oral tumors (Supplementary Fig. 2). Some studies combined melanomas from nasal and oral cavities into a head and neck category, resulting in obfuscation of the differences between these two entities29,35,46,47.
Alterations of GNAS, GNAQ, and GNA11 are widespread in different cancer types including uveal melanoma48,49. In this cohort, driver mutations in genes encoding G-protein α subunits were rare; canonical GNA11 p.Q209L substitution was detected only in one tumor.
Pathologic activation of PI3K/AKT/mTOR pathway may occur due to other than BRAF/RAS/NF1 genetic alterations such as mutations in genes encoding pathway components and regulators50,51. This study documented mutations in 14% (13/90) of SNTMMs in a wide array of genes involved in PI3K/AKT/mTOR pathway including Class I catalytic and regulatory molecules (PIK3CA, PIK3CG, PIK3R2), Class II molecule PIK3C2B, AKT1, mTOR, TSC2 and PTEN. Previous studies reported similar mutations in both cutaneous and mucosal melanomas including SNTMM14,16,29,47. In this study, only three PI3K mutants harbored canonical melanoma driver (NRAS or NF1) mutations.
The TP53 tumor suppressor gene is the most mutated gene in solid tumors52. However, TP53 mutations are rare in melanoma53. In this study, TP53 mutations and TP53 locus deletions were identified in 6% and 15% of SNTMM, respectively. Previous studies reported a similar frequency of TP53 mutations in SNTMMs and head and neck mucosal tumors13,14,15,17,29. Nevertheless, nuclear accumulation of TP53 was common, suggesting dysfunction of the TP53 pathway. In a subset of SNTMMs, nuclear expression of MDM2, a TP53 regulatory protein, mirrored nuclear accumulation of TP53. Either TP53 mutations or overexpression of MDM2 can lead to loss of TP53 tumor suppressor function54.
WNT/β-catenin signaling pathway controls a variety of biological cell processes55. Inactivating mutations of APC or activating mutations of CTNNB1, genes encoding pathway components, have been reported in cancer56. APC and CTNNB1 mutations are rare in melanoma and often co-occur with other drivers57. In this study, three SNTMMs harbored mutations in CTNNB1 exon 3 phosphorylation sites for GSK-3β (glycogen synthase kinase 3 beta) or casein kinase-1. A translocation of β-catenin to the nucleus associated with the activation of WNT/β-catenin pathway was documented in all three mutants as previously reported in other tumors58. CTNNB1 mutations affecting exon 3 hot-spots have been reported at an early stage of the tumorigenesis and have a transforming potential59. No canonical melanoma drivers were identified in one tumor harboring CTNNB1 p.S45 deletion.
A splicing factor 3b subunit 1 (SF3B1) gene encodes a subunit of the spliceosome factor 3b, a core component of ribonucleoprotein complex (spliceosome) responsible for removing introns from precursor mRNA. SF3B1 hot-spot (p.R625 and p.K666) mutations have been associated with diverse alternative splicing events and reported in uveal and mucosal (anorectal, vulvovaginal, esophageal) melanomas16,60,61,62. More recent studies identified SF3B1 mutations in head and neck melanomas including sinonasal tumors9,29. In this cohort, no SF3B1 mutations were detected in 21 tumors evaluated by Ion AmpliSeq™ CC panel. A review of reported mutants (Supplementary Table 8) suggested low (<3%) frequency of SF3B1 hot-spot mutations in SNTMMs.
Alteration of the insulin-like growth factor (IGF) axis has been implicated in carcinogenesis63. A recent study reported IGF2R mutations in 32% (13/41) of mucosal melanomas, including head and neck tumors47. In the current study, no mutations affecting IGF1R or IGF2R were found in 21 tumors analyzed with a comprehensive cancer panel. Also, previous NGS studies on mucosal melanomas including head and neck tumors failed to identify mutations affecting components of IGF-axis14,15,16,17,42.
Sequence variations in genes encodings RTKs, chromatin regulatory factors and histone proteins and genes encoding components of different DNA repair pathways (Fig. 2) were identified in a subset of SNTMMs. A biological significance of these changes remains undetermined because somatic versus germline nature could not be clearly established. However, a low VAF value (< 40%) might strongly suggest former in some cases (Supplemental Database, sheet A). Presence of pathogenic germline variants such as MET p.T1010I and MET p.R988C (reported in this study) may enhance constitutive protein tyrosine phosphorylation causing tumorigenicity in-vitro and in-vivo as reported in breast and lung cancer, respectively64,65. Recently published study showed that tumors harboring pathogenic germline variants often displayed a loss of heterozygosity or biallelic event with somatic mutations affecting the same residue66. Unfortunately, this investigation could not address such issues because a scope was limited by the absence of tumor matching normal tissue and inadequate quality of nucleic acids obtained from archival FFPE tissue blocks.
TERT alterations, from single nucleotide mutations to complex rearrangements, have been reported in different cancers including melanoma67. In this study, mutations in two TERTp mutation hot spots were detected in 9% of analyzed tumors. However, PCR amplification of these region yielded no amplification products in 14 cases with well-preserved nucleic acids. This could be attributed to the alteration of TERTp sequence. TERT fusion transcripts, NUP50(i1)::TERT(e2) and CNOT4(e2)::TERT(e2) were detected in two SNTMMs. Previously, TERT fusions involving various partners were identified in different cancers including oral melanoma (www.cbioportal.org). TERT alterations reported in this study were detected in both Triple-WT tumors and SNTMM driven by NRAS mutations. Thus, they could act as the primary driving force as reported in non-translocation related sarcomas and clear cell sarcoma of kidney, or as a secondary driver accelerating tumor progression as reported in aggressive meningioma and metastatic Leydig cells tumor67,68,69,70,71,72.
Oncogenic gene fusion involving ALK, BRAF, MET, NTRK and ROS-1 have been detected predominantly in younger patients in a subset of cutaneous melanomas including Spitzoid and acral tumors67,73,74. Such fusions appear to be extremely rare in mucosal melanomas, although a few has been reported including a FGFR3::TACC3 fusion in a case of SNTMM74,75,76. Of the 40 tumors analyzed in this study, none showed fusion gene transcripts reported in cutaneous melanomas. However, a DCAF7(e1)::PRKCA(e3) fusion, predicted to be out-of-frame, was found in one case. Activation of PRKCA through oncogenic fusion was reported in melanocytic tumors including acral melanoma77,78. Chimeric ADCK4::NUMBL fusion transcripts, most likely a product of cis-splicing between adjacent genes, were found in eight cases including 7 BRAF/RAS-WT tumors. Cis-fusion transcripts were detected in various tumor and normal tissues and implicated in fundamental cellular mechanisms79,80. However, a role of ADCK4::NUMBL fusion transcripts in SNTMM is not known.
In summary, this study documented the dominant role of NRAS oncogenesis in SNTMM and alterations of the key components and regulators of Ras-MAPK signaling pathway such as SPRED1 in a subset of BRAF/RAS-WT tumors. Also, presence of low-frequency mutations affecting KIT receptor tyrosine kinase, G-protein α subunits, TP53-, and WNT-, pathways indicate a complexity of the molecular mechanisms underlying the pathogenesis and progression of sinonasal melanoma.
Data availability
Data available upon request subjected to NIH policies and procedures.
References
Lucke A. Die melanotischen Geschwulste. Die Lehre von de Geschwulsten in anatomischet und lkinische Beziehung. In: Itha F, Billroth T, eds. Handbuch Der Allgemeinen und Speziellen Chirurgie, Erlangen, Band 2, Abteil 1, Seite 244 (1869)
Thompson LD, Wieneke JA, Miettinen M. Sinonasal tract and nasopharyngeal melanomas: a clinicopathologic study of 115 cases with a proposed staging system. Am J Surg Pathol 27, 594-611(2003)
Marcus DM, Marcus RP, Prabhu RS, Owonikoko TK, Lawson DH, Switchenko J, et al. Rising incidence of mucosal melanoma of the head and neck in the United States. J Skin Cancer 2012, 231693 (2012)
Amit M, Na’ara S, Hanna EY. Contemporary Treatment Approaches to Sinonasal Mucosal Melanoma. Curr Oncol Rep 20, 10 (2018)
Chraybi M, Abd Alsamad I, Copie-Bergman C, Baia M, André J, Dumaz N, et al. Oncogene abnormalities in a series of primary melanomas of the sinonasal tract: NRAS mutations and cyclin D1 amplification are more frequent than KIT or BRAF mutations. Hum Pathol 44, 1902-1911 (2013)
Öztürk Sari Ş, Yilmaz İ, Taşkin OÇ, Narli G, Şen F, Çomoğlu Ş, et al. BRAF, NRAS, KIT, TERT, GNAQ/GNA11 mutation profile analysis of head and neck mucosal melanomas: a study of 42 cases. Pathology 49, 55-61 (2017)
Toscano de Mendonça UB, Cernea CR, Matos LL, Monteiro de Araujo Lima RR. Analysis of KIT gene mutations in patients with melanoma of the head and neck mucosa: a retrospective clinical report. Oncotarget 9, 22886-22894 (2018)
Turri-Zanoni M, Medicina D, Lombardi D, Ungari M, Balzarini P, Rossini C, et al. Sinonasal mucosal melanoma: Molecular profile and therapeutic implications from a series of 32 cases. Head Neck 35, 1066-1077 (2013)
Wroblewska JP, Mull J, Wu CL, Fujimoto M, Ogawa T, Marszalek A, et al. SF3B1, NRAS, KIT, and BRAF Mutation; CD117 and cMYC Expression; and Tumoral Pigmentation in Sinonasal Melanomas: An Analysis With Newly Found Molecular Alterations and Some Population-Based Molecular Differences. Am J Surg Pathol 43, 168-177 (2019)
Zebary A, Jangard M, Omholt K, Ragnarsson-Olding B, Hansson J. KIT, NRAS and BRAF mutations in sinonasal mucosal melanoma: a study of 56 cases. Br J Cancer 109, 559-564 (2013)
Colombino M, Paliogiannis P, Cossu A, De Re V, Miolo G, Botti G, et al. BRAF Mutations and Dysregulation of the MAP Kinase Pathway Associated to Sinonasal Mucosal Melanomas. J Clin Med 8, 1577 (2019)
Ablain J, Xu M, Rothschild H, Jordan RC, Mito JK, Daniels BH, et al. Human tumor genomics and zebrafish modeling identify SPRED1 loss as a driver of mucosal melanoma. Science 362, 1055-1060 (2018)
Amit M, Tam S, Abdelmeguid AS, Roberts DB, Takahashi Y, Raza SM, et al. Mutation status among patients with sinonasal mucosal melanoma and its impact on survival. Br J Cancer 116, 1564-1571 (2017)
Freiberger SN, Morand GB, Turko P, Wager U, Dummer R, Hüllner M, et al. Morpho-Molecular Assessment Indicates New Prognostic Aspects and Personalized Therapeutic Options in Sinonasal Melanoma. Cancers (Basel) 11, 1329 (2019)
Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, et al. Whole-genome landscapes of major melanoma subtypes. Nature 545, 175-180 (2017)
Hintzsche JD, Gorden NT, Amato CM, Kim J, Wuensch KE, Robinson SE, et al. Whole-exome sequencing identifies recurrent SF3B1 R625 mutation and comutation of NF1 and KIT in mucosal melanoma. Melanoma Res 27, 189-199 (2017)
Newell F, Kong Y, Wilmott JS, Johansson PA, Ferguson PM, Cui C, et al. Whole-genome landscape of mucosal melanoma reveals diverse drivers and therapeutic targets. Nat Commun 10, 3163 (2019)
Stout LA, Kassem N, Hunter C, Philips S, Radovich M, Schneider BP. Identification of germline cancer predisposition variants during clinical ctDNA testing. Sci Rep 11, 13624 (2021)
Chang MH, Kuo YJ, Ho CY, Kuan EC, Lan MY. Metastatic Tumors of the Sinonasal Cavity: A 15-Year Review of 17 Cases. J Clin Med 8, 539 (2019)
Röhrich M, Koelsche C, Schrimpf D, Capper D, Sahm F, Kratz A, et al. Methylation-based classification of benign and malignant peripheral nerve sheath tumors. Acta Neuropathol 131, 877-887 (2016)
Argani P, Aulmann S, Illei PB, Netto GJ, Ro J, Cho HY, et al. A distinctive subset of PEComas harbors TFE3 gene fusions. Am J Surg Pathol 34, 1395-1406 (2010)
Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, Hu LL. ERK/MAPK signaling pathway and tumorigenesis. Exp Ther Med 19, 1997-2007 (2020)
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature 417, 949-954 (2002)
The Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 161, 1681-1696 (2015)
Omholt K, Grafström E, Kanter-Lewensohn L, Hansson J, Ragnarsson-Olding BK. KIT pathway alterations in mucosal melanomas of the vulva and other sites. Clin Cancer Res 17, 3933-3942 (2011)
Maldonado-Mendoza J, Ramírez-Amador V, Anaya-Saavedra G, Ruíz-García E, Maldonado-Martínez H, Fernández Figueroa E, et al. CD117 immunoexpression in oral and sinonasal mucosal melanoma does not correlate with somatic driver mutations in the MAPK pathway. J Oral Pathol Med 48, 382-388 (2019)
Quek C, Rawson RV, Ferguson PM, Shang P, Silva I, Saw RPM, et al. Recurrent hotspot SF3B1 mutations at codon 625 in vulvovaginal mucosal melanoma identified in a study of 27 Australian mucosal melanomas. Oncotarget 10, 930-941 (2019)
Mikkelsen LH, Maag E, Andersen MK, Kruhøffer M, Larsen AC, Melchior LC, et al. The molecular profile of mucosal melanoma. Melanoma Res 30, 533-542 (2020)
Cosgarea I, Ugurel S, Sucker A, Livingstone E, Zimmer L, Ziemer M, et al. Targeted next generation sequencing of mucosal melanomas identifies frequent NF1 and RAS mutations. Oncotarget 8, 40683-40692 (2017)
Krauthammer M, Kong Y, Bacchiocchi A, Evans P, Pornputtapong N, Wu C, et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat Genet. 2015;47:996-1002.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401-404 (2012)
Cirenajwis H, Lauss M, Ekedahl H, Törngren T, Kvist A, Saal LH, et al. NF1-mutated melanoma tumors harbor distinct clinical and biological characteristics. Mol Oncol 11, 438-451 (2017)
Lorenzo C, McCormick F. SPRED proteins and their roles in signal transduction, development, and malignancy. Genes Dev 34, 1410-1421 (2020)
Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol 24, 4340-4346 (2006)
Beadling C, Jacobson-Dunlop E, Hodi FS, Le C, Warrick A, Patterson J, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res 14, 6821-6828 (2008)
Satzger I, Schaefer T, Kuettler U, Broecker V, Voelker B, Ostertag H, et al. Analysis of c-KIT expression and KIT gene mutation in human mucosal melanomas. Br J Cancer 99, 2065-2069 (2008)
Torres-Cabala CA, Wang WL, Trent J, Yang D, Chen S, Galbincea J, et al. Correlation between KIT expression and KIT mutation in melanoma: a study of 173 cases with emphasis on the acral-lentiginous/mucosal type. Mod Pathol 22,1446-1456 (2009)
Handolias D, Hamilton AL, Salemi R, Tan A, Moodie K, Kerr L, et al. Clinical responses observed with imatinib or sorafenib in melanoma patients expressing mutations in KIT. Br J Cancer 102, 1219-1223 (2010)
Abysheva SN, Iyevleva AG, Efimova NV, Mokhina YB, Sabirova FA, Ivantsov AO, et al. KIT mutations in Russian patients with mucosal melanoma. Melanoma Res 21, 555-559 (2011)
Kong Y, Si L, Zhu Y, Xu X, Corless CL, Flaherty KT, et al. Large-scale analysis of KIT aberrations in Chinese patients with melanoma. Clin Cancer Res17, 1684-1691 (2011)
Furney SJ, Turajlic S, Stamp G, Nohadani M, Carlisle A, Thomas JM, et al. Genome sequencing of mucosal melanomas reveals that they are driven by distinct mechanisms from cutaneous melanoma. J Pathol 230, 261-269 (2013)
Hodi FS, Corless CL, Giobbie-Hurder A, Fletcher JA, Zhu M, Marino-Enriquez A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol 31, 3182-3190 (2013)
Aulmann S, Sinn HP, Penzel R, Gilks CB, Schott S, Hassel JC, et al. Comparison of molecular abnormalities in vulvar and vaginal melanomas. Mod Pathol 27, 1386-1393 (2014)
Lyu J, Song Z, Chen J, Shepard MJ, Song H, Ren G, et al. Whole-exome sequencing of oral mucosal melanoma reveals mutational profile and therapeutic targets. J Pathol 244, 358-366 (2018)
Ma X, Wu Y, Zhang T, Song H, Jv H, Guo W, et al. The clinical significance of c-Kit mutations in metastatic oral mucosal melanoma in China. Oncotarget 8, 82661-82673 (2017)
Heppt MV, Roesch A, Weide B, Gutzmer R, Meier F, Loquai C, et al. Prognostic factors and treatment outcomes in 444 patients with mucosal melanoma. Eur J Cancer 81, 36-44 (2017)
Iida Y, Salomon MP, Hata K, Tran K, Ohe S, Griffiths CF, et al. Predominance of triple wild-type and IGF2R mutations in mucosal melanomas. BMC Cancer 18,1054 (2018)
Parish AJ, Nguyen V, Goodman AM, Murugesan K, Frampton GM, Kurzrock R. GNAS, GNAQ, and GNA11 alterations in patients with diverse cancers. Cancer 124, 4080-4089 (2018)
Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med 363, 2191-2199 (2010)
Stahl JM, Cheung M, Sharma A, Trivedi NR, Shanmugam S, Robertson GP. Loss of PTEN promotes tumor development in malignant melanoma. Cancer Res 63, 2881-2890 (2003)
Shull AY, Latham-Schwark A, Ramasamy P, Leskoske K, Oroian D, Birtwistle MR, et al. Novel somatic mutations to PI3K pathway genes in metastatic melanoma. PLoS One 7, e43369 (2012)
Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol 2, a001008 (2010)
Hocker T, Tsao H. Ultraviolet radiation and melanoma: a systemic review and analysis of reported sequence variants. Hum Mutat 28, 588 (2007)
Shangary S, Wang S. Targeting the MDM2-p53 interaction for cancer therapy. Clin Cancer Res 14, 5318-5324 (2008)
Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell 149, 1192-1205 (2012)
Polakis P. Wnt signaling in cancer. Cold Spring Harb Perspect Biol 4, a008052 (2012)
Reifenberger J, Knobbe CB, Wolter M, Blaschke B, Schulte KW, Pietsch T, et al. Molecular genetic analysis of malignant melanomas for aberrations of the WNT signaling pathway genes CTNNB1, APC, ICAT and BTRC. Int J Cancer 100, 549-556 (2002)
Kim G, Kurnit KC, Djordjevic B, Singh C, Munsell MF, Wang WL et al. Nuclear β-catenin localization and mutation of the CTNNB1 gene: a context-dependent association. Mod Pathol 31, 1553-1559 (2018)
Gao C, Wang Y, Broaddus R, Sun L, Xue F, Zhang W. Exon 3 mutations of CTNNB1 drive tumorigenesis: a review. Oncotarget 9, 5492-5508 (2017)
Lasota J, Kowalik A, Felisiak-Golabek A, Zięba S, Waloszczyk P, Masiuk M, et al. Primary malignant melanoma of esophagus: clinicopathologic characterization of 20 cases including molecular genetic profiling of 15 tumors. Mod Pathol 32, 957-966 (2019)
Harbour JW, Roberson ED, Anbunathan H, et al. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet 45, 133-135 (2013)
Kong Y, Krauthammer M, Halaban R. Rare SF3B1 R625 mutations in cutaneous melanoma. Melanoma Res 24, 332-334 (2014)
Pollak MN, Schernhammer ES, Hankinson SE. Insulin-like growth factors and neoplasia. Nat Rev Cancer 4, 505-518 (2004)
Liu S, Meric-Bernstam F, Parinyanitikul N, Wang B, Eterovic AK, Zheng X, et al. Functional consequence of the MET-T1010I polymorphism in breast cancer. Oncotarget 6, 2604-2614 (2015)
Lawrence RE, Salgia R. MET molecular mechanisms and therapies in lung cancer. Cell Adh Migr 4, 146-152 (2010)
Huang KL, Mashl RJ, Wu Y, Ritter DI, Wang J, Oh C, et al. Pathogenic Germline Variants in 10,389 Adult Cancers. Cell 173, 355-370 (2018)
Yuan X, Larsson C, Xu D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: old actors and new players. Oncogene 38, 6172-6183 (2019)
Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun 5, 4846 (2014)
Delespaul L, Lesluyes T, Pérot G, Brulard C, Lartigue L, Baud J, et al. Recurrent TRIO Fusion in Nontranslocation-Related Sarcomas. Clin Cancer Res 23, 857-867 (2017)
Karlsson J, Lilljebjörn H, Holmquist Mengelbier L, Valind A, Rissler M, et al. Activation of human telomerase reverse transcriptase through gene fusion in clear cell sarcoma of the kidney. Cancer Lett 357, 498-501 (2015)
Juratli TA, Silverman IM, Shankar GM, Tummala SS, Ely HA, Christiansen JH, et al. TERT rearrangements to identify a subset of aggressive meningiomas. J Clin Oncol 36 (Suppl 15), e14028 (2018)
Kruslin B, Gatalica Z, Hes O, Xiu J, Florento E, Swensen J. TERT gene fusions characterize a subset of metastatic Leydig cell tumours. Ann Oncol 30 (Suppl 5), 981P (2019)
Wiesner T, He J, Yelensky R, Esteve-Puig R, Botton T, Yeh I, et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun 5, 3116 (2014)
Ross JS, Wang K, Chmielecki J, Gay L, Johnson A, Chudnovsky J, et al. The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int J Cancer 138, 881-890 (2016)
Kim HS, Jung M, Kang HN, Kim H, Park CW, Kim SM, et al. Oncogenic BRAF fusions in mucosal melanomas activate the MAPK pathway and are sensitive to MEK/PI3K inhibition or MEK/CDK4/6 inhibition. Oncogene 36, 3334-3345 (2017)
Lee J, Lee J, Hong SD, Jang KT, Lee SJ. FGFR3-TACC3: a novel gene fusion in malignant melanoma. Preci Future Med 2, 71-75 (2018)
Bahrami A, Lee S, Wu G, Kerstetter J, Rahvar M, Li X, et al. Pigment-Synthesizing Melanocytic Neoplasm With Protein Kinase C Alpha (PRKCA) Fusion. JAMA Dermatol 152, 318-322 (2016)
Yeh I, Jorgenson E, Shen L, Xu M, North JP, Shain AH, et al. Targeted Genomic Profiling of Acral Melanoma. J Natl Cancer Inst 111, 1068-1077 (2019)
Singh S, Qin F, Kumar S, Elfman J, Lin E, Pham LP, et al. The landscape of chimeric RNAs in non-diseased tissues and cells. Nucleic Acids Res 48, 1764-1778 (2020)
Zhang Y, Gong M, Yuan H, Park HG, Frierson HF, Li H. Chimeric transcript generated by cis-splicing of adjacent genes regulates prostate cancer cell proliferation. Cancer Discov 2, 598-607 (2012)
Funding
Federal Govorment, National Cancer Institute.
Author information
Authors and Affiliations
Contributions
Conception and design: MC, JL, MM Case selection, acquisition of clinical data: AA, WB, MD, ID, AH, SI, EI-Ś, HK, JK, JL, MM, MM, MM, RP, MP-S, AS, ST, LDRT, Immunohistochemistry: MK, JL, YL, MM, MN, Molecular genetics: MC, KH, AK, KK, AK, JL, MS, BW, Writing, review, and/or revision of the manuscript: MC, AK, JL, MM, LDRT, BW, Study supervision: JL, MM.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
Data/specimens used in this study have been de-identified by contributors prior to the investigation.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chłopek, M., Lasota, J., Thompson, L.D.R. et al. Alterations in key signaling pathways in sinonasal tract melanoma. A molecular genetics and immunohistochemical study of 90 cases and comprehensive review of the literature. Mod Pathol 35, 1609–1617 (2022). https://doi.org/10.1038/s41379-022-01122-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-022-01122-7
This article is cited by
-
Top 10 Basaloid Neoplasms of the Sinonasal Tract
Head and Neck Pathology (2023)
-
Translocations and Gene Fusions in Sinonasal Malignancies
Current Oncology Reports (2023)
-
PRAME Staining in Sinonasal Mucosal Melanoma: A Single-Center Experience
Head and Neck Pathology (2022)
