
Abstract
Rhabdomyomas are benign tumors with skeletal muscle differentiation that are broadly divided into cardiac and extracardiac types. The latter demonstrate a predilection for head and neck and genital locations and are further subclassified into adult-type rhabdomyoma (ATRM), fetal-type rhabdomyoma (FTRM) and genital rhabdomyoma (GRM). Most extracardiac rhabdomyomas that arise in paratesticular tissues have a somewhat distinctive morphology and have been termed sclerosing rhabdomyomas (SRM). Therefore, we hypothesized that these tumors may harbor recurrent genetic alterations. In this study, we assessed 15 paratesticular rhabdomyomas (11 initially classified as SRM, 2 cellular FTRM and 2 ATRM) using massively parallel DNA and RNA sequencing. Five of 14 successfully sequenced cases harbored a novel H3C2 p.K37I mutation (4 SRM and 1 ATRM). This mutation replaced a highly conserved lysine residue that is a target for epigenetic modifications and plays a role in regulation of DNA replication. Moreover, 4 tumors (2 cellular FTRM, 1 case initially diagnosed as SRM and 1 ATRM) had complex copy number profiles characterized by numerous chromosome-level and arm-level copy number gains, consistent with a ploidy shift. Rereview of the SRM with copy number gains demonstrated that it was significantly more cellular and had a more prominent fascicular architecture than the rest of the SRMs included in this series. Therefore, it was retrospectively reclassified as a cellular FTRM. In conclusion, this study demonstrated that paratesticular rhabdomyomas harbor recurrent somatic H3C2 p.K37I mutations and ploidy shifts.
Similar content being viewed by others

Introduction
Rhabdomyomas are a group of benign soft tissue tumors which exhibit skeletal muscle differentiation and are broadly divided into cardiac and extracardiac types1,2,3. By definition, the former arise in the heart of infants and children1, while the latter have a predilection for head and neck and genital locations and may affect both pediatric and adult patients2,3. Extracardiac rhabdomyomas are further subclassified based on their histologic appearances into adult-type rhabdomyoma (ATRM), fetal-type rhabdomyoma (FTRM) and genital rhabdomyoma (GRM)2.
Cardiac rhabdomyomas are a major component of the tuberous sclerosis complex4,5 and harbor predominantly TSC2 mutations6,7,8. Significantly less is known about the molecular and syndromic associations of extracardiac rhabdomyomas. FTRM and ATRM have occasionally been reported in patients with the tuberous sclerosis complex9, Gorlin syndrome10 and Birt-Hogg-Dubé syndrome11. However, a few prior molecular analyses of extracardiac rhabdomyomas have not identified recurrent oncogenic variants12,13.
Extracardiac rhabdomyomas that arise in paratesticular tissues often have distinctive morphologic features and appear to represent a distinct histologic subtype, which has been referred to as sclerosing rhabdomyoma (SRM)14. We hypothesized that these neoplasms may harbor a common underlying genetic alteration. In this study we evaluated the histopathologic and molecular features of a series of paratesticular rhabdomyomas, including cases classified as SRM, ATRM and FTRM.
Materials and methods
This study was performed with approval of the Institutional Review Board of Brigham and Women’s Hospital (BWH; Partners Health Care/ Mass General Brigham).
Accrual of the cases and histopathologic evaluation
Institutional databases and personal consultation files (CDMF, JKM, TMU) were queried to identify paratesticular rhabdomyomas. Cases with archival formalin-fixed paraffin-embedded tissue (FFPE; blocks or slides) were further selected for inclusion. Slides (H&E and immunohistochemistry) were retrieved and reviewed by the submitting authors at the corresponding institutions. Subsequently, representative slides were centrally reviewed at BWH (AMA and CDMF) to gather pertinent histopathologic information, including histologic subtype, growth pattern, entrapment of normal structures, and number of mitoses per 10 high-power fields (HPFs). Data on patient age, tumor location/laterality and gross tumor size were obtained from the original pathology reports and consultation letters. Six ATRM unselected for site of origin with archival FFPE material available were also retrieved and sequenced (DNA only) for comparison.
DNA sequencing (OncoPanel)
Massively parallel DNA sequencing was performed using a clinically validated 447-gene panel (OncoPanel, Center for Advanced Molecular Diagnostics; BWH; Supplementary Table) as previously described15,16. Briefly, FFPE neoplastic tissue was manually dissected from 1 to 8 unstained/unbaked slides using a corresponding H&E-stained section marked by a pathologist (AMA) as a guide. Samples were dissected to attempt to enrich for 20% tumor cellularity, but samples with lower tumor contents (~5–10%) were also accepted (see Discussion below). DNA was extracted with a commercial kit (Qiagen, Valencia, CA) according to the manufacturer’s recommendation and subsequently sheared by sonication. Libraries were prepared with a commercial kit (TruSeq LT library preparation kit; Illumina, San Diego, California) using a target input of 200 ng/mL of DNA (threshold 100 ng/mL) per sample. Sequences of interest were captured using custom-designed hybridization probes (Agilent SureSelect; Agilent Technologies, Santa Clara, CA) and sequenced on an Illumina HiSeq 2500 platform (Illumina, San Diego, CA). Deconvolution of batched samples, alignment of sequences, and calling and annotation of genetic variants were performed with a validated institutional informatic pipeline15,16,17. In-house developed algorithms were used to evaluate mismatch repair status and mutational signatures (UV, smoking, APOBEC, POLE)18. Genetic variants present at a frequency ≥ 0.1% in the gnomAD database (Broad Institute) were automatically filtered out to reduce contamination with germline variants. All reported variants were further assessed for biological relevance and actionability by a molecular pathologist (LMS).
RNA sequencing (gene fusion panel)
RNA sequencing for detection of gene fusions was performed at the molecular pathology laboratory of the University of Toronto as previously described by Dickson et al.19. In summary, tumor areas were marked by a pathologist (AMA) and dissected manually from 1 to 5 FFPE tissue sections (unstained-unbaked slides). Extraction of RNA was performed with a commercial kit (ExpressArt FFPE Clear RNA Ready kit; Amsbio, Cambridge, MA) and total RNA was assessed (Qubit RNA HS Assay kit, Thermofisher Scientific, Mississauga, ON, Canada). Libraries were prepared (TruSight RNA Fusion Panel; Illumina) with 20–100 ng of RNA per sample and sequenced with 76 bp paired-end reads on a MiSeq platform (Illumina, San Diego, CA). Samples were multiplexed (8 samples per flow cell), generating a total of ~3 million reads per sample. Sequencing data was assessed with two different informatic pipelines: STAR aligner with Manta fusion caller through the Illumina Local Run Manager (v.1.3.0) and BOWTIE2 alignment with the JAFFA fusion caller20,21. Fusions were considered stochastic if they: 1) had been previously identified in the context of another well-known driver in the institutional database or 2) did not result in an open frame or 3) were nonexonic or 4) had only a few supporting reads (low confidence calls).
Results
Clinicopathologic description of the cases
Fifteen paratesticular rhabdomyomas from 15 individual patients collected between 2006 and 2022 were included in this study, including 7 cases previously published by our group (cases 1, 2, 4, 5, 11, 12, and 15)14. Patients were mostly young adults, with a median age of 27 years (range: 19–72 years). The median tumor size was 4.2 cm (range 1.2–12 cm). Tumor sites included epididymis in 4 cases, spermatic cord in 3 cases, and paratesticular/scrotal tissue, not further specified in the remaining 8 cases. The laterality was left in 8 cases and right in 7 cases.
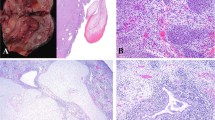
The demographic, histopathologic and immunohistochemical features of the cases are summarized in Table 1. Based on morphology, 11 cases were initially classified as SRM (including the 7 cases previously published by Jo et al.)14, 2 as cellular FTRM and 2 as ATRM. Briefly, SRMs were characterized by sparse bundles of mature polygonal and elongated rhabdomyoblasts with copious cytoplasm embedded in an abundant sclerotic collagenous stroma (Fig. 1). Lymphoplasmacytic infiltrates and/or lymphoid aggregates were invariably present in SRM, which resulted in a relatively low ratio of neoplastic to nonneoplastic nuclei. Cellular FTRMs demonstrated higher cellularity than SRMs, with a mixture of primitive spindle cells and more differentiated rounded and elongated rhabdomyoblasts arranged in short fascicles (Fig. 2). The two ATRM consisted of sheets of plump polygonal rhabdomyoblasts with occasional cytoplasmic filamentous (“jackstraw”) inclusions and minimal intervening stroma (Fig. 3). Scattered microscopic foci of infarction were present in one of these ATRM, and the remaining one exhibited several small cysts measuring up to 0.7 cm in greatest dimension. The more differentiated rhabdomyoblasts had similar cytomorphology across the different subtypes of rhabdomyoma. Specifically, they had relatively large nuclei, conspicuous nucleoli, abundant eosinophilic cytoplasm with variably noticeable striations and frequent intracytoplasmic inclusions. Tumor necrosis was not identified, and mitotic activity was consistently below 1 mitotic figure per 10 HPF in all cases. Immunohistochemical stains for desmin, MyoD1, Myf4, and fast myosin were positive in all cases in which they were performed (12/12, 6/6, 1/1, and 2/2, respectively).

A–D Case 1 (A), case 2 (B), case 4 (C) and case 5 (D) demonstrate the characteristic histopathologic features of sclerosing rhabdomyomas. These tumors are typically hypocellular, with bundles of well-differentiated rhabdomyoblasts embedded in an abundant collagenous stroma that contains lymphoplasmacytic infiltrates and lymphoid aggregates. The four cases illustrated in this figure harbored a recurrent H3C2 p.K37I mutation.

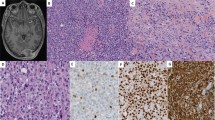
A. Case 6 was hypercellular with a mixture of spindle cells and mature rhabdomyoblasts arranged in bundles and fascicles. B Case 8 was hypercellular and consisted predominantly of mature rhabdomyoblasts arranged in short fascicles. These cases had a copy number profile characterized by multiple chromosome-level and arm-level copy number gains.

A, B Case 3 consisted of hypercellular sheets of polygonal rhabdomyoblasts with minimal intervening stroma. Filamentous (“jackstraw”) inclusions were present in scattered rhabdomyoblasts (inset, panel B). This case harbored a H3C2 p.K37I mutation.
DNA and RNA sequencing results
Fourteen cases underwent successful DNA sequencing and one failed due to excessive DNA fragmentation (Table 2 and Supplementary Fig. 1). Ten cases with additional FFPE tissue available were submitted for RNA sequencing. Of these, 4/10 were sequenced successfully and 6/10 failed extraction (4/6) or sequencing (2/6).
DNA sequencing identified a novel H3C2 p.K37I mutation in 5/14 (36%) cases, including 4 SRMs and 1 ATRM. The mechanisms of mutation included an A to T transversion at nucleotide position 110 (cases 1, 2, 4, and 5), and a dinucleotide inversion (case 3). Case 2 also harbored a frameshift FANCE variant of likely germline origin based on the variant allele frequency. One additional SRM (case 7) harbored a frameshift MSH6 variant without evidence of concurrent mismatch repair deficiency. A frameshift mononucleotide deletion was identified in BRIP1 in 1 of the 2 cellular FTRMs, while the remaining 7 cases with interpretable results (5 SRM, 1 cellular FTRM, and 1 ATRM) were mutationally silent.
Chromosome-level and arm-level copy number changes were present in 4 tumors (cases 6–9), including the 2 cellular FTRMs, 1 lesion initially classified as SRM, and 1 ATRM (see discussion below). The copy number profile of these 4 neoplasms was characterized by multiple copy number gains that spanned large chromosomal regions (not shown in Table 2), chromosome arms and entire chromosomes, consistent with ploidy shifts.
Only 1 gene fusion (MON1A::CIC; case 3) was detected by RNA sequencing (but not by DNA sequencing). This was a low-confidence call present in the context of a concurrent H3C2 p.K37I mutation, and was considered stochastic (i.e., nonpathogenic).
DNA sequencing of additional ATRM
Given the molecular findings in paratesticular rhabdomyomas, additional ATRM unselected for site of origin were sequenced for comparison. FFPE tissue was available to attempt DNA sequencing on 6 ATRM (all from head and neck locations) from 4 patients. Four tumors from 2 patients (including 3 metachronous ATRM from a single patient) were sequenced successfully. The 3 metachronous tumors from a single patient harbored a truncating frameshift FLCN variant (p.E297Afs*25) present at a frequency highly suggestive of a germline event (~50%), without definite evidence of loss of heterozygosity. Additional pathogenic mutations were not identified in any of these 4 ATRM arising in head and neck locations. Of note, H3C2 was manually reviewed in all cases and the p.K37I variant was not identified.
Discussion
Extracardiac rhabdomyomas are benign mesenchymal neoplasms exhibiting skeletal muscle differentiation that are subclassified into ATRM, FTRM, and GRM according to their clinicopathologic features. ATRMs usually arise in the head and neck of adult patients, and their histology is characterized by sheets of plump polygonal rhabdomyoblasts with minimal intervening stroma. FTRMs affect mostly infants and young children and have a predilection for the head and neck in general, and for the postauricular region in particular. Their histology spans a spectrum that includes hypocellular tumors with predominantly immature spindle cells embedded in an abundant myxoid stroma on one end, and hypercellular tumors with more differentiated rhabdomyoblasts arranged in fascicles on the other. GRM typically arise in the vagina and vulva of adult women and consist of a submucosal proliferation of variably mature rhabdomyoblasts, often mimicking embryonal rhabdomyosarcoma.
Most paratesticular rhabdomyomas demonstrate distinctive histologic features, characterized by scattered bundles of well-differentiated rhabdomyoblasts embedded in an abundant collagenous stroma14. These tumors, termed SRM, have also been reported in the pelvic floor of an adult woman22, suggesting that they are predominantly, but not exclusively, paratesticular. Moreover, individual examples of paratesticular FTRM, ATRM and GRM have been documented in pediatric and adult patients23,24,25,26,27. This study expands our prior experience with paratesticular rhabdomyomas, confirming that although SRM morphology is dominant, FTRM and ATRMs also occur in this location.
Extracardiac rhabdomyomas may recur locally if incompletely resected2, but no recurrences have been observed in paratesticular cases14. Paratesticular rhabdomyomas may mimic malignant tumors, including embryonal rhabdomyosarcoma, alveolar soft part sarcoma (ATRM), spindle cell/sclerosing rhabdomyosarcoma (SRM) and dedifferentiated liposarcoma with heterologous rhabdomyoblastic differentiation. Like SRM, sclerosing rhabdomyosarcoma exhibits a prominent collagenous stroma with hyalinization but typically lacks large rhabdomyoblasts28. Embryonal rhabdomyosarcoma and spindle cell rhabdomyosarcoma are moderately-to-highly cellular lesions composed predominantly of spindle and round cells, with histologic appearances that overlap with those of FTRM29. The large polygonal cells of ATRM are morphologically similar to rhabdomyoblasts occasionally seen in dedifferentiated liposarcoma and to the neoplastic cells of alveolar soft part sarcoma30,31. Therefore, awareness of the morphologic spectrum of paratesticular rhabdomyomas is necessary to avoid a misdiagnosis of malignancy.
Unlike cardiac rhabdomyomas, which have a well-demonstrated association with the tuberous sclerosis complex and TSC1/2 mutations32, extracardiac rhabdomyomas have not been consistently associated with any specific molecular alteration. FTRMs have been described in patients with Gorlin syndrome10, and PTCH1 mutations have been identified in this tumor type previously33. ATRM has been reported in the context of Birt–Hogg–Dubé syndrome11, a disorder caused by FLCN mutations. However, molecular analyses of additional series and individual cases of extracardiac rhabdomyomas have not identified pathogenic genetic variants in these neoplasms12,13. In this study, 3 metachronous ATRM arising in head and neck locations of a single patient harbored a truncating FLCN variant of likely germline origin, without unequivocal evidence of loss of heterozygosity. Per clinical notes, the patient had a history of multiple episodes of spontaneous pneumothorax treated with pleurodesis at a young age, highly suggestive of Birt-Hogg-Dubé syndrome.
The present study found that, overall, 9/14 (64%) paratesticular rhabdomyomas sequenced successfully had characteristic molecular findings that appear to be mutually exclusive. More specifically, a recurrent H3C2 p.K37I variant and multiple chromosomal gains consistent with ploidy shifts were identified in 5/14 (36%) and 4/14 (29%) cases, respectively. The H3C2 p.K37I variant was the only recurrent finding in paratesticular SRM, without morphologic differences between H3C2-mutant and H3C2-wild type SRM (Fig. 4), and multiple chromosomal gains were the only finding in paratesticular FTRM. Based on the small number of cases assessed herein, paratesticular ATRM may harbor either of these molecular alterations. None of the 4 typical ATRM arising in head and neck locations harbored the H3C2 p.K37I variant or multiple chromosomal gains. These results seem to suggest that the molecular alterations identified in this study might be enriched in ATRM arising in paratesticular tissues, akin to BAP1 and KIT mutations in uveal and acral melanoma, respectively34,35. However, the conclusions that can be drawn from this analysis are limited because 3 extra-scrotal neoplasms were metachronous lesions from a single patient. Further studies are necessary to determine the frequency of the H3C2 p.K37I mutation and ploidy shifts in ATRM and to explore the presence of alternative drivers in H3C2 wild-type SRM. Because SRMs have abundant stroma, variably prominent lymphoplasmacytic infiltrates and tumor cells that are significantly more voluminous than the intermingled nonneoplastic cells, it is likely that tumor cellularity was overestimated in a subset of cases, limiting the detection of H3C2 p.K37I mutations in additional SRM. Importantly, no paratesticular tumors in this series had known syndromic associations or harbored pathogenic genetic variants in TSC1/2, PTCH1, FLCN or SUFU, all of which are included in our panel.

A Case 10. B Case 13 are represented in the images.
H3C2 codes for histone 3.1, an isoform of histone 3 that is deposited during DNA synthesis and repair36. In the S phase, the histone chaperone CAF-1 promotes assembly of histone octamers that contain this particular isoform of histone 3. In contrast, in other phases of the cell cycle, the histone chaperone HIRA promotes assembly of histone octamers that contain histone 3.336. This explains why histone 3.3 is enriched in differentiated tissues with low proliferation rates36,37. The K37 residue of histone 3 is highly conserved across species and is a known target for epigenetic modifications. In the budding yeast S. cerevesiae, monomethylation of this residue by Set1/2 promotes initiation of DNA replication at canonical replication origins and prevents the initiation of replication at non-canonical sites38. In S. pombe, methylation of K37 by Set7 increases during gametogenesis and is required for normal gamete formation, while disruption of K37 methylation results in abnormal immature gametes (i.e., spores)39. Mutations that replace the conserved K37 residue of histone 3.3A and histone 3.3B (H3F3A and H3F3B) have been reported previously in giant cell tumor of bone40. However, this is the first time that a mutation involving K37 of histone 3.1 (H3C2) has been identified in a human tumor, adding to the repertoire of genetic variants seen in so-called oncohistones41.
Interestingly, the CNV profiles of the 2 cellular-type FTRMs, 1 case initially classified as SRM (case 7) and 1 ATRM (case 9) were characterized by multiple arm-level and chromosome-level copy number gains, consistent with a ploidy shift. Given these results, the slides of these cases were re-reviewed by two of the authors (AMA and CDMF). Comparative re-assessment of these cases demonstrated that the tumor originally classified as SRM was significantly more cellular and had a more prominent fascicular arrangement than the other SRMs included in the series (Fig. 5). Therefore, this tumor was retrospectively reclassified as a cellular-type FTRM with sclerotic stroma. Interestingly, the ATRM with multiple chromosomal gains exhibited histologic features that were typical of this variant, including the presence of “jackstraw” cytoplasmic inclusions. This suggests that paratesticular ATRM comprise a group of tumors with heterogeneous molecular alterations, including H3C2 p.K37I and ploidy shifts.

A, B Case 7 was originally diagnosed as a sclerosing rhabdomyoma. DNA sequencing identified multiple chromosome-level and arm-level copy number gains in this case, which were not seen in other sclerosing rhabdomyomas. In light of the molecular findings, slides were re-reviewed demonstrating that this tumor was significantly more cellular and had a more prominent fascicular arrangement than the rest of the sclerosing rhabdomyomas (compare to Fig. 1). Therefore, this tumor was reclassified as cellular fetal rhabdomyoma with sclerotic stroma.
The main shortcoming of this study is its relatively small sample size, which is almost inevitable with these rare lesions. Moreover, the series includes old archival cases (>10 years) with relatively low cellularity, which may have limited the detection of the H3C2 mutation in additional cases. All mutation-negative cases were manually reviewed at codon 37, reducing the risk of false negative results due to variants present below the threshold of the bioinformatics filters. Despite the shortcomings mentioned above, the present series represent the largest compilation of paratesticular rhabdomyomas and the first multiplatform molecular evaluation of this entity to date.
In conclusion, the present study has demonstrated that paratesticular rhabdomyomas with SRM morphology harbor a recurrent H3C2 p.K37I mutation, which replaces a highly conserved lysine residue that is subject to epigenetic modifications and seems to be involved in regulation of DNA replication38. In contrast, paratesticular rhabdomyomas with cellular-FTRM morphology harbor multiple arm-level and chromosome-level copy number gains. ATRM seem to represent an intermediate group with molecular alterations that overlap those seen in SRM and FTRM.
Data availability
The data generated in this study are available from the corresponding author upon reasonable request.
References
Fenoglio, J. J., MCAllister, H. A. & Ferrans, V. J. Cardiac rhabdomyoma: A clinicopathologic and electron microscopic study. Am J Cardiol 38, 241–251 (1976).
Willis, J., Abdul-Karim, F. W. & di Sant’Agnese, P. A. Extracardiac rhabdomyomas. Semin Diagn Pathol 11, 15–25 (1994).
Di Sant’Agnese, P. A. & Knowles, D. M. Extracardiac rhabdomyoma: a clinicopathologic study and review of the literature. Cancer 46, 780–789 (1980).
Sciacca, P., Giacchi, V., Mattia, C., Greco, F., P Smilari, Betta, P. et al. Rhabdomyomas and tuberous sclerosis complex: our experience in 33 cases. BMC Cardiovasc Disord 14, 66 (2014).
Kocabaş, A., Ekici, F., Cetin, I. İ., Emir, S., Demir, H. A., Arı, M. E. et al. Cardiac rhabdomyomas associated with tuberous sclerosis complex in 11 children: presentation to outcome. Pediatr Hematol Oncol 30, 71–79 (2013).
Al Kindi, H. N., Ibrahim, A. M., Roshdy, M., Abdelghany, B. S., Yehia, D., Masoud, A. N. et al. Clinical, cellular, and molecular characterisation of cardiac rhabdomyoma in tuberous sclerosis. Cardiol Young 31, 1297–1305 (2021).
Chen, L., Jiang, Y. & Wang, J. Fetal cardiac rhabdomyoma due to paternal mosaicism of TSC2: A case report. Medicine (Baltimore) 99, e21949 (2020).
Chen, C.-P., Su, Y.-N., Hung, C.-C., Shih, J.-C. & Wang, W. Novel mutation in the TSC2 gene associated with prenatally diagnosed cardiac rhabdomyomas and cerebral tuberous sclerosis. J Formos Med Assoc 105, 599–603 (2006).
Elawabdeh, N., Sobol, S., Blount, A. C. & Shehata, B. M. Unusual presentation of extracardiac fetal rhabdomyoma of the larynx in a pediatric patient with tuberous sclerosis. Fetal Pediatr Pathol 31, 43–47 (2013).
Hettmer, S., Teot, L. A., Kozakewich, H., Werger, A. M., Davies, K. J., Fletcher, C. D. M. et al. Myogenic tumors in nevoid Basal cell carcinoma syndrome. J Pediatr Hematol Oncol 37, 147–149 (2015).
Balakumar, R., Farr, M. R. B., Fernando, M., Jebreel, A., Ray, J. & Sionis, S. Adult-Type Rhabdomyoma of the Larynx in Birt-Hogg-Dubé Syndrome: Evidence for a Real Association. Head Neck Pathol 13, 507–511 (2019).
De la Iglesia Niveyro, P. X., Pandolfi, J., Jauk, F., Kreindel, T. & Lobos, P. Prostatic Rhabdomyoma in a Toddler: A Case Report With Molecular Characterization. Pediatr Dev Pathol 25, 203-206 (2022).
Schoolmeester, J. K., Xing, D., Keeney, G. L. & Sukov, W. R. Genital Rhabdomyoma of the Lower Female Genital Tract: A Study of 12 Cases With Molecular Cytogenetic Findings. Int J Gynecol Pathol 37, 349–355 (2018).
Jo, V. Y., Reith, J. D., Coindre, J. M. & Fletcher, C. D. M. Paratesticular rhabdomyoma: a morphologically distinct sclerosing variant. Am J Surg Pathol 37, 1737–1742 (2013).
Sholl, L. M., Do, K., Shivdasani, P., Cerami, E., Dubuc, A. M., Kuo, F. C. et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight 1, e87062 (2016).
Garcia, E. P., Minkovsky, A., Jia, Y., Ducar, M. D., Shivdasani, P., Gong, X. et al. Validation of OncoPanel: A Targeted Next-Generation Sequencing Assay for the Detection of Somatic Variants in Cancer. Arch Pathol Lab Med 141, 751–758 (2017).
Abo, R. P., Ducar, M., Garcia, E. P., Thorner, A. R., Rojas-Rudilla, V., Lin, L. et al. BreaKmer: detection of structural variation in targeted massively parallel sequencing data using kmers. Nucleic Acids Res 43, e19 (2015).
Papke, D. J., Nowak, J. A., Yurgelun, M. B., Frieden, A., Srivastava, A., Lindeman, N. I. et al. Validation of a targeted next-generation sequencing approach to detect mismatch repair deficiency in colorectal adenocarcinoma. Mod Pathol 31, 1882–1890 (2018).
Dickson, B. C. & Swanson, D. Targeted RNA sequencing: A routine ancillary technique in the diagnosis of bone and soft tissue neoplasms. Genes Chromosomes Cancer 58, 75–87 (2019).
Liu, S., Tsai, W.-H., Ding, Y., Chen, R., Fang, Z., Huo, Z. et al. Comprehensive evaluation of fusion transcript detection algorithms and a meta-caller to combine top performing methods in paired-end RNA-seq data. Nucleic Acids Res 44, e47 (2016).
Chen, X., Schulz-Trieglaff, O., Shaw, R., Barnes, B., Schlesinger, F., Källberg, M. et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222 (2016).
Quijano Moreno, S. L., Lozano Salazar, A. D., Del Mar Berenguel Ibáñez, M., Reina Duarte, Á. & Gonzales Campora, R. “Sclerosing” Pelvic Floor Rhabdomyoma. Int J Surg Pathol 24, 159–162 (2016).
Kurzrock, E. A., Busby, J. E. & Gandour-Edwards, R. Paratesticular rhabdomyoma. J Pediatr Surg 38, 1546–1547 (2003).
Tanda, F., Rocca, P. C., Bosincu, L., Massarelli, G., Cossu, A. & Manca, A. Rhabdomyoma of the tunica vaginalis of the testis: a histologic, immunohistochemical, and ultrastructural study. Mod Pathol 10, 608–611 (1997).
Maheshkumar, P. & Berney, D. M. Spermatic cord rhabdomyoma. Urology 56, 331 (2000).
Leite, K. R. M., Dantas, K. O. F., de Azevedo, L. S. & Camara-Lopes, L. H. Paratesticular rhabdomyoma. Ann Diagn Pathol 10, 239–240 (2006).
Han, Y., Qiu, X., Li, Q., Han, Y., Lin, X., Zhang, Q. et al. Epididymis rhabdomyoma: a case report and literature review. Diagn Pathol 7, 47 (2012).
Folpe, A. L., McKenney, J. K., Bridge, J. A. & Weiss, S. W. Sclerosing rhabdomyosarcoma in adults: report of four cases of a hyalinizing, matrix-rich variant of rhabdomyosarcoma that may be confused with osteosarcoma, chondrosarcoma, or angiosarcoma. Am J Surg Pathol 26, 1175–1183 (2002).
Leuschner, I., Newton, W. A., Schmidt, D., Sachs, N., Asmar, L., Hamoudi, A. et al. Spindle cell variants of embryonal rhabdomyosarcoma in the paratesticular region. A report of the Intergroup Rhabdomyosarcoma Study. Am J Surg Pathol 17, 221–230 (1993).
Shimada, S., Ishizawa, T., Ishizawa, K., Kamada, K. & Hirose, T. Dedifferentiated liposarcoma with rhabdomyoblastic differentiation. Virchows Arch 447, 835–841 (2005).
Folpe, A. L. & Deyrup, A. T. Alveolar soft-part sarcoma: a review and update. J Clin Pathol 59, 1127–1132 (2006).
Zhen, L., Yang, Y.-D., He, Y., Pan, M., Han, J., Yang, X. et al. Prenatal genetic diagnosis of cardiac rhabdomyoma: A single-center experience. Eur J Obstet Gynecol Reprod Biol 249, 7–10 (2020).
Hettmer, S., Teot, L. A., van Hummelen, P., MacConaill, L., Bronson, R. T., Dall’Osso, C. et al. Mutations in Hedgehog pathway genes in fetal rhabdomyomas. J Pathol 231, 44–52 (2013).
Karlsson, J., Nilsson, L. M., Mitra, S., Alsén, S., Shelke, G. V., Sah, V. R. et al. Molecular profiling of driver events in metastatic uveal melanoma. Nat Commun 11, 1894 (2020).
Torres-Cabala, C. A., Wang, W.-L., Trent, J., Yang, D., Chen, S., Galbincea, J. et al. Correlation between KIT expression and KIT mutation in melanoma: a study of 173 cases with emphasis on the acral-lentiginous/mucosal type. Mod Pathol 22, 1446–1456 (2009).
Tagami, H., Ray-Gallet, D., Almouzni, G. & Nakatani, Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 116, 51–61 (2004).
Wunsch, A. M. & Lough, J. Modulation of histone H3 variant synthesis during the myoblast-myotube transition of chicken myogenesis. Dev Biol 119, 94–99 (1987).
Santos-Rosa, H., Millán-Zambrano, G., Han, N., Leonardi, T., Klimontova, M., Nasiscionyte, S. et al. Methylation of histone H3 at lysine 37 by Set1 and Set2 prevents spurious DNA replication. Mol Cell 81, 2793-2807.e8 (2021).
Shen, Y., Mevius, D. E. H. F., Caliandro, R., Carrozzini, B., Roh, Y., Kim, J. et al. Set7 Is a H3K37 Methyltransferase in Schizosaccharomyces pombe and Is Required for Proper Gametogenesis. Structure 27, 631-638.e8 (2019).
Kervarrec, T., Collin, C., Larousserie, F., Bouvier, C., Aubert, S., Gomez-Brouchet, A. et al. H3F3 mutation status of giant cell tumors of the bone, chondroblastomas and their mimics: a combined high resolution melting and pyrosequencing approach. Mod Pathol 30, 393–406 (2017).
Nacev, B. A., Feng, L., Bagert, J. D., Lemiesz, A. E., Gao, J., Soshnev, A. A. et al. The expanding landscape of “oncohistone” mutations in human cancers. Nature 567, 473–478 (2019).
Acknowledgements
The authors would like to thank the following pathologists who kindly submitted cases included in this study: Dr. Jose Manuel Lopes, Porto, Portugal; Dr. Emilia Gottberg, Lund, Sweden; Dr. Pehr Rissler, Lund, Sweden; Dr. Julia Solares, Caceres, Spain; Dr. Jung Wan, Cortland, NY, USA; Dr. Paul Sindler, Penrith, Australia; Dr. Foteini Karasavvidou, Larissa, Greece; Dr. Alex Chang, Nashville, TN, USA; Dr. Susan Prendeville, Cork, Ireland; Dr. Alae Yasees, Sacramento, CA, USA; Dr. John D. Reith, Gainesville, FL, USA; Dr. Kathleen Coleman, Valdosta, GA, USA; Dr. Mauricio Palau, Bogota, Colombia; Dr. Kathleen Coleman, Valdosta, GA, USA.
Funding
Support for RNA sequencing has been provided by the Panov 2 Research Fund (Dr. BCD).
Author information
Authors and Affiliations
Contributions
Concept: AMA and JKMcK; design and coordination: AMA and CDM. Fletcher; analysis of the sequencing data: LMS and BD; correlation of histopathologic and molecular results: AMA, LMS, and CDM. Fletcher; contribution of cases: CDM. Fletcher, JKMcK, TMU, and KC; manuscript draft and figures: AMA; intellectual contributions and manuscript editing: all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study was performed with approval of the Institutional Review Board of Brigham and Women’s Hospital (BWH; Partners Health Care/ Mass General Brigham).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Acosta, A.M., McKenney, J.K., Sholl, L.M. et al. Molecular assessment of paratesticular rhabdomyomas demonstrates recurrent findings, including a novel H3C2 p.K37I mutation. Mod Pathol 35, 1921–1928 (2022). https://doi.org/10.1038/s41379-022-01134-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-022-01134-3
