
Abstract
A small subset of male germ cell tumors (GCT) demonstrates overgrowth of histologic components that resemble somatic malignancies (e.g., sarcoma, carcinoma). The presence of so-called “somatic-type” malignancies (SM) in GCT has been associated with chemotherapy-resistance and poor clinical outcomes in prior studies. However, the molecular characteristics of these tumors remain largely undescribed. In this study, we performed a multi-platform molecular analysis of GCTs with SM diagnosed in 36 male patients (primary site: testis, 29 and mediastinum, 7). The most common histologic types of SM were sarcoma and embryonic-type neuroectodermal tumor (ENT, formerly known as “PNET”), present in 61% and 31% of cases, respectively. KRAS and TP53 mutations were identified by DNA sequencing in 28% of cases each, with enrichment of TP53 mutations in mediastinal tumors (86%). Gains in the short arm of chromosome 12 were seen in 91% of cases, likely reflecting the presence of isochromosome 12p. Numerous copy number changes indicative of widespread aneuploidy were found in 94% of cases. Focal homozygous deletions and amplifications were also detected, including MDM2 amplifications in 16% of cases. Sequencing of paired samples in 8 patients revealed similar mutational and copy number profiles in the conventional GCT and SM components. Oncogenic gene fusions were not detected using RNA sequencing of SM components from 9 cases. DNA methylation analysis highlighted the distinct methylation profile of SM components that sets them apart from conventional GCT components. In conclusion, GCT with SM are characterized by widespread aneuploidy, a distinct epigenetic signature and the presence of mutations that are otherwise rare in testicular GCT without SM. The similarity of the mutational and DNA methylation profiles of different histologic types of SM suggests that the identification of SM components could be more important than their precise histologic subclassification, pending confirmation by further studies.
Similar content being viewed by others

Introduction
Testicular germ cell tumors (GCTs), the most common cancer type in men aged 14 to 44 years, are broadly grouped into seminoma and non-seminoma categories for clinical management1. Non-seminomatous tumors comprise mixed GCTs (which may include components of seminoma) and GCTs with pure non-seminoma histology1,2. Most male GCTs are thought to arise from testicular germ cell precursors that undergo neoplastic transformation. A smaller subset of GCTs arising in extra-testicular sites such as the mediastinum, retroperitoneum and brain is believed to be derived from primordial germ cells that fail to migrate to the gonads during embryonic development3.
From a genetic perspective, testicular GCTs are characterized by whole-genome duplication, a low mutational burden and relative gains of sequences present in the short arm of chromosome 12, usually in the form of an isochromosome 12p [i(12p)] or regional amplification events4. Recurrent gain-of-function mutations of KIT, KRAS, and NRAS are seen almost exclusively in seminoma4. Extra-testicular GCTs also demonstrate gains of sequences of the short arm of chromosome 12 such as i(12p)5 but, unlike testicular GCTs6,7, they often harbor TP53 mutations8,9.
Surgery followed by platinum-based chemotherapy in qualifying cases achieves cure rates of ~95% and ~80–90% in patients with localized and metastatic testicular GCTs, respectively10,11. However, ~10% of patients develop resistance to chemotherapy and have a dismal prognosis despite aggressive treatment12,13. A subset of GCT resistant to systemic treatment is characterized by overgrowth of histologic components resembling somatic-type malignancies (SM), including sarcomas, carcinomas and, rarely, myeloid neoplasms14,15,16,17,18.
In GCTs, SM components are believed to arise from teratoma in most cases14,17; however, a smaller subset appears to be derived from other non-seminomatous components, such as yolk sac tumor16,17. Detection of i(12p) by fluorescence in situ hybridization (FISH)19,20 or quantitative PCR21 can be useful in cases of SM that are not associated with conventional GCT components, especially in the metastatic setting. Prior studies have described the clinicopathologic characteristics of GCT with SM14,15,16,17, but the molecular correlates of so-called “somatic transformation” remain poorly understood. In this study, we performed a multi-institutional, multi-platform molecular assessment of testicular and extratesticular male GCTs with SM.
Materials and methods
Accrual of cases, demographic and clinicopathologic data
This research was performed with approval of the Institutional Review Board of Brigham and Women’s Hospital (BWH; MGB Insight 4.0, protocol #2021P002289).
The informatic databases of the participating institutions and the personal consultation files of the authors were queried to identify testicular and extratesticular GCTs with SM diagnosed in male patients. One case (case #3) was previously published as a case report22. Slides were retrieved and reviewed by the participating pathologists at their corresponding institutions. Cases with overgrowth of SM (defined as SM components spanning >1 low power field) and archival formalin-fixed paraffin embedded (FFPE) tissue available for molecular studies were included. All available slides of BWH cases and selected slides of the remaining cases were centrally reviewed at BWH by two of the authors (NW and AMA). Demographic and clinicopathologic data were obtained by review of pathology reports, clinical notes and summarized information submitted by the authors.
DNA sequencing (OncoPanel)
Samples were assessed using a solid tumor next-generation sequencing panel (OncoPanel; 447 genes) as previously described by our group in validation studies23,24. Tumor content was enriched (target tumor cellularity ≥20%) by manual dissection of FFPE tissue sections (5 µm) using corresponding H&E slides marked by a pathologist (NW and AMA) as a guide. In samples with areas of conventional GCT and SM amenable to separation by manual dissection, these components were differentially dissected and sequenced in parallel (see “Paired DNA sequencing of SM and conventional GCT components” section below). In these cases, SM and conventional GCT were dissected either from different blocks (when possible) or from well-separated areas present in the same block. Otherwise, tissue was dissected to enrich for SM components. DNA was extracted with a commercial kit (Qiagen, Valencia, CA) following the manufacturer’s recommendations and subsequently sheared by sonication. Whenever possible, 200 ng of DNA per sample were used for library preparation. Samples that yielded less than 100 ng of DNA were rejected. Libraries were constructed using a commercial kit (TruSeq LT library preparation kit; Illumina, San Diego, California) and the sequences of interest were captured by hybridization to a set of custom-designed DNA probes (Agilent SureSelect; Agilent Technologies, Santa Clara, CA). Sequencing by synthesis was performed on an Illumina HiSeq 2500 sequencer (Illumina, San Diego, CA). A clinically validated informatics pipeline was used for sample deconvolution, alignment of the generated sequences, variant calling and annotation23,24,25. Mutational signatures (POLE, APOBEC, smoking, UV) and mismatch repair status26,27 were assessed by algorithms developed at the Center for Advanced Molecular Diagnostics (CAMD, BWH). Genomic variants present at a frequency >0.1% in the gnomAD database (Broad Institute) were automatically excluded to avoid contamination with germline events. The reported variants were evaluated and tiered for actionability by a molecular pathologist (LMS).
RNA sequencing (fusion panel)
Targeted RNA sequencing was performed on cases with additional FFPE tissue available (#1, #3, #7, #12, #20, #21, #23, #31, and #33) at Mount Sinai Hospital, University of Toronto as previously described28. Tumor areas were marked by a pathologist (NW and AMA) and dissected manually from on 1–5 unstained FFPE sections. Commercial kits were used for total RNA extraction (ExpressArt FFPE Clear RNA Ready kit; Amsbio, Cambridge, MA) and assessment (Qubit RNA HS Assay kit, Thermofisher Scientific, Mississauga, ON, Canada). A minimum of 20 ng and a maximum of 100 ng of RNA per sample was used for library preparation (TruSight RNA Fusion Panel; Illumina). Samples were multiplexed (8 per flow cell) and sequenced as 76-bp paired-end reads on a MiSeq platform (Illumina, San Diego, CA), resulting in ~3 million reads per sample. Sequencing results were analyzed using two different informatic pipelines: STAR aligner with Manta fusion caller through the Illumina Local Run Manager (v.1.3.0) and BOWTIE2 alignment with the JAFFA fusion caller29,30. Fusions were considered clinically or biologically relevant (i.e., not stochastic) if they had an open reading frame, had not been identified previously in the context of another well-known driver in our database, and had enough supporting reads.
Methylation profile analysis
DNA methylation profiling was performed as previously reported31. Briefly, FFPE tissue was dissected manually to enrich for tumor cells for DNA extraction. The DNA was processed for hybridization and fluorescence staining on the Infinium MethylationEPIC (850 k) BeadChip array (Illumina, San Diego, USA) according to the manufacturer’s instructions. The arrays were scanned in the Illumina iScan microarray scanner. Raw idat files were analyzed in R version and raw intensities were processed with the Bioconductor R package Minfi32. Each sample was assessed for quality by mean detection p-value (p < 0.05). Samples were normalized by quantile normalization, followed by removal of probes that failed in one or more samples (detection p < 0.01), and of probes with single-nucleotide polymorphisms. Beta-values were calculated with the default offset value 100 recommended for Illumina assays. Differential methylation analysis was based on comparing samples with conventional GCT histologies and samples with SM components. After fitting linear models with limma (3.48.3)33, top differentially methylated probes were selected based on adjusted p-values (p < 0.05). A Beta value lower than 0.2 or higher than 0.8 was defined as hypomethylation or hypermethylation, respectively. A heatmap based on the hierarchical clustering of the top 1000 differentially methylated probes was generated using ComplexHeatmap (2.8.0)34.
Results
General description of the series and tumor histology
GCT with SM components from 36 patients were included in the study. The primary tumor was testicular in 29 patients and mediastinal in the remaining 7 patients (Table 1). The average patient age was 34 years (range 13 to 69 years), without differences between patients with testicular and mediastinal primaries.
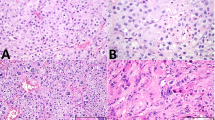
The most common type of somatic-type malignancy in our series was sarcoma (22/36, 61%, Fig. 1), including 12 cases of unclassified spindle and/or epithelioid sarcoma (12/22, 55%), 9 cases of rhabdomyosarcoma (9/22, 41%), and 1 case of angiosarcoma (1/22, 5%). Embryonic type neuroectodermal tumor (ENT) was present in 11 cases (11/36, 31%). Carcinoma and nephroblastoma were each found in 2 patients (2/36, 6%). Pancreatoblastoma and small blue round cell tumor, NOS were also present (1/36, 3%). Two cases (2/36, 6%) showed multiple SM components (case #19: ENT and nephroblastoma; case #20: rhabdomyosarcoma, ENT and unclassified sarcoma).

Selected examples of somatic-type malignancy from the series: A unclassified sarcoma, B rhabdomyosarcoma, C angiosarcoma, and D Embryonic type neuroectodermal tumor (ENT, formerly known as “PNET”).
In addition to the SM components, 28 cases also contained one or more conventional GCT component (28/36, 78%), including teratoma (22/36, 61%), yolk sac tumor (12/36, 33%), embryonal carcinoma (9/36, 25%), and seminoma (7/36, 19%). Fifteen of these cases (15/36, 42%) had more than one conventional GCT component.
Panel DNA sequencing and fusion panel RNA sequencing of enriched SM components
Thirty-six SM arising in GCT underwent DNA sequencing, including cases in which SM components were either enriched or separated from concurrent conventional GCT components by manual dissection (see “Paired DNA sequencing of SM and conventional GCT components” below). The tumors selected for next-generation sequencing included 15 testicular primaries, 3 mediastinal primaries, 13 retroperitoneal metastases and 5 metastases to the lung. Four specimens (cases # 8, 9, 19, and 36) were obtained after chemotherapy and the remaining tumors were treatment-naïve or their treatment status was unknown (see Table 1). Two sequenced tumors (cases #9 and #36) were late recurrences, resected 14 and 4 years after initial treatment, respectively.
Single nucleotide variant analysis was performed on 32 cases (32/36, 89%, Fig. 2). The remaining 4 cases were excluded because they failed pre-established quality control metrics. The most frequently mutated genes in this series were KRAS and TP53, with mutations detected in 9 cases each (9/32, 28%). KMT2D was mutated in 5 cases (5/32, 16%). A pathogenic KIT mutation was detected in 1 case (1/32, 3%). TP53 mutations were significantly enriched in GCTs of mediastinal origin (86% vs 12%). Of note, primary SM, metastatic SM and the different histologic types of SM had similar mutational profiles (Fig. 2).

The position of cases from left to right correspond to the case numbers in Table 1. Abbreviations: KRAS Kirsten rat sarcoma viral oncogene, TP53 tumor protein p53 gene, KMT2D histone-lysine N-methyltransferase 2D, MDM2 murine double minute 2, CNV copy number variant, NTRK neurotrophic tyrosine receptor kinase, CDKN2A/B cyclin dependent kinase inhibitor 2A/B, EP300 E1A binding protein p300 gene, CRKL Crk-like protein gene, MAPK1 mitogen-activated protein kinase 1, JAZF1 juxtaposed with another zinc finger protein 1, MYC myelocytomatosis oncogene, RB1 retinoblastoma protein 1, FGFR1 fibroblast growth factor receptor 1, MET MET proto-oncogene.
Despite overall low sequencing quality in 5 cases, focal gains/amplifications were identified on manual review. Copy number gains and/or amplification of the short arm of chromosome 12 were seen in 32 cases (32/35, 91%). In addition, 30 SM (30/31, 97%) harbored numerous arm-level gains and losses with frequent widespread loss of heterozygosity, indicative of widespread aneuploidy. Focal cancer-relevant gene amplifications were seen in 10 cases (10/33, 30%), including (but not restricted to) MDM2 in 5 cases (5/33, 15%), MYC in 2 cases (2/31, 6%), and NTRK, JAZF, FGFR1 and MET in 1 case each (1/31, 3%). Homozygous (i.e., “deep”) deletions of RB1, CDKN2A/B, MRAE11 and EP300 genes were detected in 1 case each (1/31, 3%). Pathogenic structural variants such as in-frame fusions of genes covered by the panel (e.g., EWSR1::FLI1) were not identified in any of the cases analyzed herein.
Enriched SM components from a subset of 9 tumors underwent gene fusion analysis by RNA sequencing. None of the 6 cases (4 unclassified sarcomas, 1 rhabdomyosarcoma, 1 angiosarcoma) that passed quality control harbored oncogenic gene fusions.
Paired DNA sequencing of SM and conventional GCT components
Paired sequencing of SM and conventional GCT components was performed in a subset of 8 patients. More specifically, paired SM and primary testicular conventional GCT components were sequenced in 2 cases (case #4 and #25), paired SM and metastatic conventional GCT components were sequenced in 4 cases (cases #11, #18, #24, and #33), and paired SM, primary testicular conventional GCT and metastatic conventional GCT components were sequenced in 2 cases (cases #10 and #21). In one of these 8 patients, primary testicular and metastatic SM components were also sequenced in parallel (case #25). Among the remaining 7 cases, the sequenced SM components were primary in 3 cases and metastatic in 4 cases (Table 1).
In most cases, copy number analysis of the conventional GCT components revealed multiple large copy number gains and losses almost identical to those seen in the paired SM components (Fig. 3A, Supplementary Fig. 1). In 2 cases, the SM components appeared to have a more complex copy number profile than the paired conventional GCT components. However, the NGS platform used in this study is not validated for quantitative comparisons of copy number variants across samples. Of note, multiple large copy number gains and losses consistent with a high degree of aneuploidy were not a feature of conventional GCTs (i.e., without SM components) identified in our genomic database and reviewed for comparison (data not shown).

A Representative copy number variant (CNV) plots inferred from targeted NGS data of matched testicular primary GCT, metastatic conventional GCT and metastatic somatic-type malignancy (case #20). B Venn diagram of the number of private/shared single nucleotide variants (SNVs) detected in 10 (metastatic) conventional GCTs and 8 paired somatic-type malignancies.
Most pathogenic single nucleotide variants (40/53, 76%) were shared by the paired conventional GCT and SM components (Fig. 3B). Five and 8 (likely passenger) mutations were restricted to the conventional GCT and SM components (5/53, 9% and 8/53, 15%), respectively. Primary testicular and metastatic SM shared 7/8 (88%) mutations. Of note, KRAS and TP53 mutations were always shared by the paired conventional GCT and SM components.
Genome-wide methylation analysis of paired SM and conventional GCT components
Array-based DNA methylation analysis of paired SM and conventional GCT components was performed on the same specimens that underwent paired DNA sequencing (see above), except for case #33 and the metastatic SM component of case #25. In addition, paired SM and metastatic GCT components of case #8 and 4 control conventional GCT cases (2 seminomas, 1 teratoma and 1 embryonal carcinoma) that were not associated with SM were included in the DNA methylation analysis. The SM components analyzed included ENT (6 cases) and rhabdomyosarcoma (2 cases). The conventional GCT components analyzed included teratoma (7 cases), yolk sac tumor (2 cases) and seminoma (1 case), with both primary testicular and metastatic components included in 2 cases (cases #10 and #21). Supervised clustering using the top 1000 differentially methylated probes highlighted the differences between the methylation profiles of tumor components with SM and conventional GCT histology (Fig. 4). Of note, based on the analysis of the top 1000 differentially methylated probes, there were no differences between the methylation profiles of the different SM histologies (ENT and rhabdomyosarcoma) assessed herein.

ENT embryonic-type neuroectodermal tumor, YST yolk sac tumor.
Discussion
Overgrowth of histologic components resembling somatic malignancies is seen in less than 10% (2.7–8.6%) of male GCTs treated at large tertiary centers35. From a clinical perspective, GCTs with SM components are generally resistant to systemic therapy and associated with a dismal prognosis in the metastatic setting (5-year survival rate of 35%)17. This contrasts with the favorable outcomes reported in primary GCT with SM (5-year survival rate of 87%), which are comparable to those of conventional GCT17.
From a morphologic perspective, the SM components of GCTs may resemble specific types of somatic sarcomas and carcinomas, including a subset that is similar to neoplasms formerly called primitive neuroectodermal tumors (PNET)15,16,17. These PNET-like components of GCTs lack EWSR1 rearrangements36 and have recently been renamed as embryonic-type neuroectodermal tumor (ENT) to avoid misinterpretation as Ewing sarcoma (formerly known as peripheral-type PNET)37. Other subgroups of SM with epithelioid or nondescript spindle cell morphology that express keratins and glypican 3 can be classified as glandular or sarcomatoid yolk sac tumor16,38. These are often diagnosed at metastatic sites after chemotherapy and behave aggressively, especially when they demonstrate high-grade histologic features16,38,39.
Only a few molecular studies of GCT with SM had been performed to date. Assessment of these tumors with quantitative PCR demonstrated that they harbor copy number gains of sequences present in the short arm of chromosome 12, indicative of i(12p)21. As mentioned above, FISH studies using break-apart EWSR1 probes demonstrated the absence of EWSR1 rearrangements in GCT with ENT components36. However, a more comprehensive molecular interrogation of GCT with SM including paired comparative analyses of SM and conventional GCT components had not been undertaken previously.
In the present study, DNA sequencing confirmed that most testicular and extra-testicular GCT with SM harbor copy number gains involving the short arm of chromosome 12, likely reflecting the presence of i(12p) in a subset of cases. Most GCT with SM had numerous additional copy number changes consistent with widespread aneuploidy, which was not a feature of conventional GCTs from our genomic database and published studies4,37. Of note, similar copy number changes have been described in GCT resistant to chemotherapy and in sarcomatoid yolk sac tumor, another tumor type that falls within the spectrum of GCTs with “somatic transformation”9,39. DNA sequencing also revealed enrichment for TP53 mutations in GCT with SM (9/32, 28% cases in total: 3/25, 12% testicular and 6/7, 86% mediastinal). Of note, TP53 mutations are virtually nonexistent in conventional testicular GCTs sensitive to systemic therapy4,6,7. However, TP53 mutations are present in a small subset of platinum-resistant conventional testicular GCT, mostly of mediastinal origin, and a significant subset of sarcomatoid yolk sac tumors9,39. MDM2 amplification was identified in 4/25 (16%) testicular and 1/7 (14%) extra-testicular GCT with SM, being mutually exclusive with TP53 mutations in the former. In total, considering both TP53 and MDM2 variants, 13/32 (41%) cases in this series harbored p53 pathway alterations, including both primary and metastatic tumors. A significant subset of cases also harbored KRAS mutations (9/32, 28%: 6/25, 24% testicular and 3/7, 43% mediastinal). Remarkably, the copy number changes and the spectrum of driver mutations present in primary and metastatic conventional GCT components were almost identical to those seen in the corresponding (i.e., paired) SM components of the tumor. This suggests that the detection of TP53 mutations, MDM2 amplifications and widespread aneuploidy in early-stage disease might identify cases with potentially aggressive clinical behavior and resistance to chemotherapy.
A recent study17 found that the prognosis of testis-confined GCTs with SM is similar to that of testis-confined conventional GCTs, whereas metastatic SM fare significantly worse than metastatic conventional GCTs. One plausible explanation for this finding is that metastatic SM clones accumulate molecular changes that result in biologic progression. In this study, comparison of the mutational, copy number and methylation profiles of SM within an individual case (#25) as well as across different cases demonstrated that primary and metastatic SM have similar molecular characteristics. Therefore, we hypothesize that SM components of GCTs are intrinsically resistant to systemic treatment but can be cured by complete surgical resection, which would explain the striking stage-dependent differences in overall survival observed in prior studies15,17.
DNA sequencing and RNA sequencing with a panel optimized for detection of gene fusions detected no pathogenic fusions in GCT with SM resembling somatic sarcomas. Moreover, the mutational and DNA methylation profiles of different histologic types of SM analyzed herein were similar. These findings indicate that SMs arising in GCTs are biologically different from their true somatic counterparts, and they likely represent a more homogeneous entity than one would presume based on their diverse histologic appearances. Hence, our results suggest that the identification of SM components could be more important than their precise histologic subclassification, pending confirmation with further studies. Additionally, these results suggest that systemic therapy designed for the true somatic counterparts of the SM components (e.g., rhabdomyosarcoma chemotherapy for a SM with rhabdomyosarcomatous differentiation) might not be the optimal treatment in this setting40.
This study has limitations that need to be mentioned. First, paired samples were only available for a subset of cases. Second, targeted DNA and RNA sequencing panels optimized for the detection of variants in known cancer-relevant genes were performed. This approach limits the detection of novel gene alterations, but the use of clinically validated panels with well-known performance characteristics ensures that the variants identified in the study are biologically relevant. Third, paired germline studies were not performed. Although germline data is always helpful, it is not essential for the interpretation of the results of this study. Moreover, established sequencing metrics were used to infer the origin of the genetic variants41. Fourth, the number of cases is relatively small, albeit comparable to prior series of this somewhat rare entity. Fifth, carcinomas are under-represented in our cohort. A recent study17 has suggested that GCT with “somatic-type” carcinoma may behave more aggressively. Further investigations are needed to assess whether carcinomas arising in GCT harbor different molecular alterations. Despite these limitations, this is the first comprehensive multi-platform molecular characterization of a multi-institutional series of GCT with SM.
In summary, the findings of this study suggest that (1) SM arising in GCTs are biologically unrelated to their true somatic counterparts, (2) molecular features associated with “somatic-transformation” of GCTs are already present at an early disease stage (3) the different histologic subtypes of SM arising in GCT appear to be biologically similar and, therefore, their precise subclassification might not be essential, and (4) “somatic transformation” of GCTs might be driven by epigenetic events. Additional studies are needed to further elucidate the mechanistic processes that underlie “somatic transformation” in GCT.
Data availability
The data generated in this study are available from the corresponding author upon reasonable request.
References
Chen L, Albers P, Berney DM, Feldman DR, Daugaard G, Gilligan T et al. Testicular cancer. Nat. Rev. Dis. Primer 4, 1–24 (2018).
Moch H, Humphrey P, Ulbright T, Reuter V. WHO Classification of Tumours of the Urinary System and Male Genital Organs. (2016).
Oosterhuis JW, Looijenga LHJ. Human germ cell tumours from a developmental perspective. Nat. Rev. Cancer 19, 522–537 (2019).
Shen H, Shih J, Hollern DP, Wang L, Bowlby R, Tickoo SK, et al. Integrated molecular characterization of testicular germ cell tumors. Cell Rep. 23, 3392–3406 (2018).
Cin PD, Drochmans A, Moerman P, Berghe HVD. Isochromosome 12p in mediastinal germ cell tumor. Cancer Genet. Cytogenet. 42, 243–251 (1989).
Peng H-Q, Hogg D, Malkin D, Bailey D, Gallie BL, Bulbul M, et al. Mutations of the p53 gene do not occur in testis cancer. Cancer Res. 53, 3574–3578 (1993).
Schenkman NS, Sesterhenn IA, Washington L, Tong YA, Weghorst CM, Buzard GS, et al. Increased p53 protein does not correlate to p53 gene mutations in microdissected human testicular germ cell tumors. J. Urol. 154, 617–621 (1995).
Loveday C, Litchfield K, Proszek PZ, Cornish AJ, Santo F, Levy M, et al. Genomic landscape of platinum resistant and sensitive testicular cancers. Nat. Commun. 11, 2189 (2020).
Bagrodia A, Lee BH, Lee W, Cha EK, Sfakianos JP, Iyer G, et al. Genetic determinants of cisplatin resistance in patients with advanced germ cell tumors. J. Clin. Oncol. 34, 4000–4007 (2016).
Mazzone E, Knipper S, Mistretta FA, Tian Z, Palumbo C, Soulieres D, et al. Contemporary north-american population-based validation of the international germ cell consensus classification for metastatic germ cell tumors of the testis. World J. Urol. 38, 1535–1544 (2020).
Albers P, Albrecht W, Algaba F, Bokemeyer C, Cohn-Cedermark G, Fizazi K, et al. Guidelines on testicular cancer: 2015 Update. Eur. Urol. 68, 1054–1068 (2015).
Oechsle K, Kollmannsberger C, Honecker F, Mayer F, Waller CF, Hartmann JT, et al. Long-term survival after treatment with gemcitabine and oxaliplatin with and without paclitaxel plus secondary surgery in patients with cisplatin-refractory and/or multiply relapsed germ cell tumors. Eur. Urol. 60, 850–855 (2011).
Oing C, Seidel C, Bokemeyer C. Therapeutic approaches for refractory germ cell cancer. Expert Rev. Anticancer Ther. 18, 389–397 (2018).
Ulbright TM, Loehrer PJ, Roth LM, Einhorn LH, Williams SD, Clark SA. The development of non-germ cell malignancies within germ cell tumors. A clinicopathologic study of 11 cases. Cancer 54, 1824–1833 (1984).
Colecchia M, Necchi A, Paolini B, Nicolai N, Salvioni R. Teratoma with somatic-type malignant components in germ cell tumors of the testis: a clinicopathologic analysis of 40 cases with outcome correlation. Int. J. Surg. Pathol. 19, 321–327 (2011).
Magers MJ, Kao CS, Cole CD, Rice KR, Foster RS, Einhorn LH, et al. ‘Somatic-type’ malignancies arising from testicular germ cell tumors: a clinicopathologic study of 124 cases with emphasis on glandular tumors supporting frequent yolk sac tumor origin. Am. J. Surg. Pathol. 38, 1396–1409 (2014).
Hwang MJ, Hamza A, Zhang M, Tu SM, Pisters LL, Czerniak B, et al. Somatic-type malignancies in testicular germ cell tumors: a clinicopathologic study of 63 cases. Am. J. Surg. Pathol. 46, 11–17 (2022).
Taylor J, Donoghue MT, Ho C, Petrova-Drus K, Al-Ahmadie HA, Funt SA, et al. Germ cell tumors and associated hematologic malignancies evolve from a common shared precursor. J. Clin. Invest. 130, 6668–6676 (2020).
Kernek KM, Brunelli M, Ulbright TM, Eble JN, Martignoni G, Zhang S, et al. Fluorescence in situ hybridization analysis of chromosome 12p in paraffin-embedded tissue is useful for establishing germ cell origin of metastatic tumors. Mod. Pathol. 17, 1309–1313 (2004).
Kum JB, Ulbright TM, Williamson SR, Wang M, Zhang S, Foster RS, et al. molecular genetic evidence supporting the origin of somatic-type malignancy and teratoma from the same progenitor cell. Am. J. Surg. Pathol. 36, 1849–1856 (2012).
Fichtner A, Richter A, Filmar S, Gaisa NT, Schweyer S, Reis H, et al. The detection of isochromosome i(12p) in malignant germ cell tumours and tumours with somatic malignant transformation by the use of quantitative real-time polymerase chain reaction. Histopathology 78, 593–606 (2021).
Vargas AC, Grimison P, Joy C, Garrone B, Bonar F, Ghahan RM, et al. Chromosome 8 polysomy accounting for MYC over-expression in angiosarcoma arising as somatic-type malignancy in metastatic teratoma. Case report. Int. J. Surg. Pathol. 10668969211067762 (2021). https://doi.org/10.1177/10668969211067762.
Garcia EP, Minkovsky A, Jia Y, Ducar MD, Shivdasani P, Gong X, et al. Validation of OncoPanel: a targeted next-generation sequencing assay for the detection of somatic variants in cancer. Arch. Pathol. Lab. Med. 141, 751–758 (2017).
Sholl LM, Do K, Shivdasani P, Cerami E, Dubuc AM, Kuo FC, et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight 1, e87062 (2016).
Abo RP, Ducar M, Garcia EP, Thorner AR, Rojas-Rudilla V, Lin L, et al. BreaKmer: detection of structural variation in targeted massively parallel sequencing data using kmers. Nucleic Acids Res. 43, e19–e19 (2015).
Christakis AG, Papke DJ, Nowak JA, Yurgelun MB, Agoston AT, Lindeman NI, et al. Targeted cancer next-generation sequencing as a primary screening tool for microsatellite instability and lynch syndrome in upper gastrointestinal tract cancers. Cancer Epidemiol. Biomarkers Prev. 28, 1246–1251 (2019).
Papke DJ, Nowak JA, Yurgelun MB, Frieden A, Srivastava A, Lindeman NI, et al. Validation of a targeted next-generation sequencing approach to detect mismatch repair deficiency in colorectal adenocarcinoma. Mod. Pathol. 31, 1882–1890 (2018).
Dickson BC, Swanson D. Targeted RNA sequencing: A routine ancillary technique in the diagnosis of bone and soft tissue neoplasms. Genes. Chromosomes Cancer 58, 75–87 (2019).
Liu S, Tsai WH, Ding Y, Chen R, Fang Z, Huo Z, et al. Comprehensive evaluation of fusion transcript detection algorithms and a meta-caller to combine top performing methods in paired-end RNA-seq data. Nucleic Acids Res. 44, e47 (2016).
Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222 (2016).
Serrano J, Snuderl M. Whole genome DNA methylation analysis of human glioblastoma using illumina BeadArrays. in Glioblastoma: Methods and Protocols (ed. Placantonakis, D. G.) 31–51 (Springer, 2018). https://doi.org/10.1007/978-1-4939-7659-1_2.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014).
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016).
Rice KR, Magers MJ, Beck SD, Cary KC, Einhorn LH, Ulbright TM, et al. Management of germ cell tumors with somatic type malignancy: pathological features, prognostic factors and survival outcomes. J. Urol. 192, 1403–1409 (2014).
Ulbright TM, Hattab EM, Zhang S, Ehrlich Y, Foster RS, Einhorn LH, et al. Primitive neuroectodermal tumors in patients with testicular germ cell tumors usually resemble pediatric-type central nervous system embryonal neoplasms and lack chromosome 22 rearrangements. Mod. Pathol. 23, 972–980 (2010).
McIntyre A, Summersgill B, Lu YJ, Missiaglia E, Kitazawa S, Oosterhuis JW, et al. Genomic copy number and expression patterns in testicular germ cell tumours. Br. J. Cancer 97, 1707–1712 (2007).
Howitt BE, Magers MJ, Rice KR, Cole CD, Ulbright TM. Many postchemotherapy sarcomatous tumors in patients with testicular germ cell tumors are sarcomatoid yolk sac tumors: a study of 33 cases. Am. J. Surg. Pathol. 39, 251–259 (2015).
Acosta AM, Al-Obaidy KI, Sholl LM, Dickson BC, Lindeman NI, Hirsch MS, et al. Sarcomatoid yolk sac tumor harbors somatic mutations that are otherwise rare in testicular germ cell tumors. Am. J. Surg. Pathol. 5, 701–712 (2022).
Scheckel CJ, Kosiorek HE, Butterfield R, Ho TH, Hilal T. Germ cell tumors with malignant somatic transformation: a mayo clinic experience. Oncol. Res. Treat. 42, 95–100 (2019).
Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the association for molecular pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 19, 4–23 (2017).
Acknowledgements
The authors would like to thank Dr. Jon C. Aster (Pathology Chair, BWH), Mary LoGiudice-Croce (Research Administration, BWH) and Chennai Marcus (Coordinator, CAMD, BWH) for their valuable support for this research.
Funding
RNA sequencing was performed with support of the Panov 2 Research Fund (Brendan C. Dickson).
Author information
Authors and Affiliations
Contributions
Concept and coordination: AMA. Design: AMA and NW. Contribution of cases: AMA, KIA-O, NB, KC, JBG, MTI, C-SK, FM, AM, SEW, CDMF, MSH, and JLH. DNA sequencing analysis: LMS. RNA sequencing analysis: BCD. DNA methylation analysis: YY, IT, VV, and MS. Integration and interpretation of clinical, histopathologic, and molecular data: NW and AMA. Manuscript draft: NW and AMA. Figures and tables: NW. Manuscript editing: All authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This research was performed with approval of the Institutional Review Board of Brigham and Women’s Hospital (BWH; MGB Insight 4.0, protocol #2021P002289).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wyvekens, N., Sholl, L.M., Yang, Y. et al. Molecular correlates of male germ cell tumors with overgrowth of components resembling somatic malignancies. Mod Pathol 35, 1966–1973 (2022). https://doi.org/10.1038/s41379-022-01136-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-022-01136-1
This article is cited by
-
SMARCB1-Deficient Skull Base Chondrosarcoma with 12p Duplication Presenting as Somatic-Type Malignancy Arising from Metastatic Seminoma
Head and Neck Pathology (2024)
-
P53 Overexpression May Represent an Early Marker of Clinicopathologic Progression in Vasculogenic Mesenchymal Lesions of Germ Cell Tumor Origin
Virchows Archiv (2024)
-
Mediastinal high-grade vasculogenic mesenchymal tumour with seminoma: a case report and literature review
Diagnostic Pathology (2023)
-
Characterizing the mutational burden, DNA methylation landscape, and proteome of germ cell tumor-related somatic-type malignancies to identify the tissue-of-origin, mechanisms of therapy resistance, and druggable targets
British Journal of Cancer (2023)
-
Testicular neoplasms: the interrelationships of serum levels of microRNA-371a-3p (M371) and classical tumor markers with histology, clinical staging, and age—a statistical analysis
Journal of Cancer Research and Clinical Oncology (2023)
