
Abstract
Although the density of tumor-infiltrating lymphocytes (TILs) is known to be linked to prognosis in various cancers, the prognostic impact and immunologic significance of the spatial heterogeneity of TILs have been rarely investigated. In this study, CD3+ and CD8+ TILs were quantified in independent cohorts (discovery, n = 73; and external validation, n = 93) of colorectal carcinomas (CRCs) with microsatellite instability-high (MSI-H) utilizing whole-slide image analysis of CD3/CD8 immunohistochemistry. The Shannon and Simpson indices, which measure intratumoral patch-to-patch evenness of TIL densities, were used to quantitatively assess the spatial heterogeneity of TILs in each case. To uncover immune-related gene expression signatures of spatial heterogeneity-based TIL subgroups of MSI-H CRCs, representative cases were subjected to GeoMx digital spatial profiler (DSP) analysis. As expected, a low density of TILs was significantly associated with poor disease-free survival (DFS) in MSI-H CRCs. The TIL-low tumors were further classified into two subgroups based on the spatial heterogeneity of TILs: TIL-low/heterogeneity-high and TIL-low/heterogeneity-low subgroups. In both discovery and validation cohorts, the TIL-low/heterogeneity-high, TIL-low/heterogeneity-low, and TIL-high subgroups were significantly associated with poor, intermediate, and good DFS, respectively. In the DSP analysis, the TIL-low/heterogeneity-high subgroup showed higher spatial diversity in the expression of immune-related genes than that of the TIL-low/heterogeneity-low subgroup and exhibited upregulation of genes related to immune checkpoints, chemokine/cytokine receptors, and myeloid cells. TIL-low/heterogeneity-high tumors were also enriched with gene sets related to good response to immune checkpoint inhibitor therapy. In conclusion, TIL-low MSI-H CRCs are prognostically heterogeneous and can be divided into prognostically and immunologically distinct subgroups by considering the spatial heterogeneity of TILs. Our data suggest that intratumoral spatial heterogeneity of TILs can be used as a key element for clinically relevant immunologic subtyping of tumors.
Similar content being viewed by others


Introduction
The interplay between the tumor microenvironment and tumor cells critically determines tumor behavior, patient survival, and treatment response in various cancers. Notably, the tumor immune microenvironment (TIME), comprising tumor-infiltrating lymphocytes (TILs), tumor-associated macrophages, tertiary lymphoid structures (TLS), immune checkpoint expression, etc., plays a pivotal role in orchestrating tumor suppression and progression, including in colorectal carcinomas (CRCs), through immunosurveillance and immunoediting1,2,3. Among the TIME factors, the prognostic significance of TILs has been frequently investigated and confirmed in CRCs through many clinicopathologic studies based on measuring TIL density on hematoxylin and eosin (H&E)-stained or immunohistochemistry (IHC) tissue slides1,4,5,6. Although most previous studies have focused on the clinicopathologic values of TIL density in CRCs, the implications of spatial distribution patterns or heterogeneity of TILs within the whole tumor area have been poorly investigated.
Microsatellite instability-high (MSI-H), which occurs in approximately 15% of CRCs, is a molecular fingerprint of genetic defects in the DNA mismatch repair (MMR) process7. The deficiency in DNA MMR leads to an accumulation of DNA frameshift mutations that ultimately translate into increased immunogenic neoantigens6. The resulting immunogenic pressure in MSI-H CRC activates tumoricidal immunity with increased infiltration of TILs, but at the same time, the hostile TIME in MSI-H CRC is counterbalanced by immune evasion through upregulation of immune checkpoints, including PD-1, PD-L1, CTLA4, and LAG36,8. The coexistence of a high density of TILs and the high expression of immune checkpoints provides an excellent opportunity for utilizing immune checkpoint inhibitors (ICIs) in the treatment of MSI-H CRCs9.
Conceptually, most MSI-H CRCs can be regarded as highly immunogenic tumors. However, recent studies reported that TIME features in MSI-H CRCs are not monolithic but vary with different biofunctions and outcomes10,11. In line with these findings, our recent study also found that MSI-H CRCs could be divided into immune subgroups based on variable TIL/TLS profiles. These immune subgroups of MSI-H CRCs showed distinct clinicopathologic, genomic, and transcriptomic characteristics, suggesting that different treatment strategies for these subgroups may be necessary12. The optimal classification of immune subgroups of MSI-H CRCs could be valuable in precisely predicting patient survival and response to ICIs. In fact, although MSI-H CRC has been considered an ideal model for ICI treatment13, the overall response rate to ICI in MSI-H CRCs has been reported to be approximately 30–60%13,14,15,16. Thus, multi-dimensional TIL factors (i.e., not only TIL density but also TIL spatial heterogeneity) should be considered to determine the optimal classification of TIL subgroups for better prognostication in MSI-H CRCs.
Collectively, we hypothesized that immune subgrouping using TIL density only might be limited when it comes to precisely stratifying prognostic subgroups in MSI-H CRCs. Consequently, we decided to investigate the prognostic value of TIL spatial heterogeneity in MSI-H CRCs. To test this hypothesis, we mapped TILs in the whole-slide tumor area of MSI-H CRCs using a digital pathology-based quantification technique. By adopting ecological diversity indices, i.e., Shannon and Simpson indices, patch-to-patch evenness of TIL densities was assessed, and its prognostic implications were investigated. To further compare spatial context-dependent TIME gene expression profiles between TIL spatial heterogeneity subgroups, multiplex digital spatial profiler (DSP) analysis using an immuno-oncology gene panel was also conducted in selected cases in each subgroup.
Materials and methods
Tissue collection
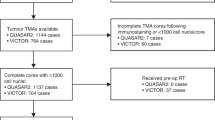
As a discovery cohort for this study, 73 MSI-H CRCs were retrospectively collected from the pathology archive of Seoul National University Hospital, Seoul, Korea as previously described12,17,18. All cases were radically resected for treatment and pathologically diagnosed as CRCs at Seoul National University Hospital between 2015 and 2017. The MSI status of each case was confirmed by fluorescence capillary electrophoresis-based DNA fragment analysis using five microsatellite markers (BAT-25, BAT-26, D5S346, D17S250, and D2S123) according to the Bethesda Guidelines19. All clinical and histopathologic data were reviewed or retrieved, as previously described, by two experienced gastrointestinal pathologists (JAL and JHK)12,17. As an external validation cohort, 93 MSI-H CRC tissues were retrospectively collected from the pathology archive of Severance Hospital, Seoul, Korea, which were radically resected for treatment and pathologically diagnosed as CRCs at Severance Hospital between 2009 and 2012. None of the patients whose MSI-H CRCs were obtained in both cohorts received preoperative neoadjuvant chemotherapy and/or radiation therapy. The demographic and clinicopathologic features of the MSI-H CRCs, including discovery and validation cohorts, are summarized in Supplementary Table S1. Briefly, according to the American Joint Committee on Cancer staging system (8th edition), 10 (13.7%), 43 (58.9%), 16 (21.9%), and 4 (5.5%) tumors in the discovery cohort and 26 (28.0%), 41 (44.1%), 23 (24.7%), and 3 (3.2%) tumors in the validation cohort were at TNM stages I, II, III, and IV, respectively (Supplementary Table S1). According to the World Health Organization CRC grading system (5th edition), 49 (67.1%) and 24 (32.9%) tumors in the discovery cohort and 74 (79.6%) and 19 (20.4%) tumors in the validation cohort were low- and high-grade tumors, respectively (Supplementary Table S1). The two cohorts had similar distributions of stages and grades (Fisher’s exact test p = 0.1 each). The hereditary subtype (Lynch or Lynch-like syndrome) of the MSI-H CRCs was determined when a tumor harbored a germline pathogenic mutation in one of the MMR genes and/or MMR IHC deficiency in tumor cells without MLH1 promoter hypermethylation. The sporadic subtype of the MSI-H CRCs was determined when a tumor exhibited both MLH1 IHC deficiency and MLH1 promoter hypermethylation. Using the electronic medical records and survival registry data, the disease-free survival (DFS) was calculated from the surgery date to that of death or tumor recurrence in each patient. The median follow-up durations in the discovery and validation cohorts were 49 and 59 months, respectively. During these periods, 13 and 14 patients experienced tumor recurrence, respectively, and six and nine patients died, respectively. The study protocol was approved by the Institutional Review Boards of Seoul National University Hospital (IRB No. 1804-036-935; and 1805-018-944) and Severance Hospital (IRB No. 4-2021-1130).
Whole-slide image analysis-based quantification of TILs
TILs in our CRC cohorts were quantified based on computational image analysis of IHC slides as previously described12,17. QuPath, a validated open-source software for digital pathology analysis20,21, was used to quantify CD3+ or CD8+ TILs on whole-section IHC slides of MSI-H CRCs. In brief, the whole tumor area on a representative tumor section IHC slide of each case was divided into 1-mm2-sized square patches by three experienced pathologists (JAL, S-YY, and MJ). The positive cell counts in a patch measured by the ‘positive cell detection’ functionality of QuPath represented the density of CD3+ or CD8+ TILs in the patch (cells/mm2). The average density of CD3+ or CD8+ TILs in each case was calculated as follows:
Quantitative assessment of the spatial heterogeneity of TILs
The intratumoral spatial heterogeneity of TIL densities was measured using the Shannon and Simpson evenness indices22,23. Both evenness indices can be calculated from their corresponding diversity indices, which are widely used approaches for quantifying ecosystem biodiversity24. They have been applied to measure the diversity or heterogeneity of cells and genes in medical research25,26,27,28,29. The Shannon and Simpson diversity indices were designed to consider richness and evenness, the two major aspects of diversity; however, the Shannon index emphasizes the richness component of diversity more than the Simpson index, whereas the Simpson index emphasizes the evenness component of diversity more than the Shannon index30. Considering the need to quantify the evenness of densities of a single species (in this study, CD3+ or CD8+ TIL) to assess TIL spatial heterogeneity inversely, we adopted the evenness indices, not the diversity indices, in the current study. We decided to use both Shannon and Simpson evenness indices because confirming which between the two indices is superior to the other in measuring spatial heterogeneity is difficult and because they could be used complementarily. We calculated the spatial heterogeneity of TILs by taking into account both the proportional abundance of TILs (pi) and total cancer area (S) as follows24,31:
In both equations, pi is the number of TILs in each patch divided by the total number of TILs, while S is the number of patches covering the tumor area. Because the difference in the number of patches in individual specimens is adjusted, the evenness indices were useful in comparing the TIL distribution between cases. If the spatial distribution of TILs is completely even and all patches contain the same number of TILs, Shannon and Simpson evenness indices would be 1. In contrast, if TILs are highly uneven and one dominant patch contains all TILs, the indices would approach 0.
GeoMx DSP analysis
The NanoString GeoMx DSP assay (NanoString Technologies, Seattle, WA, USA) was performed in 12 MSI-H CRCs selected from representative cases of the 3 TIL subgroups (TIL-low/heterogeneity-high, TIL-low/heterogeneity-low, and TIL-high subgroups) in the discovery cohort MSI-H CRCs. The detailed procedures are described elsewhere32. Briefly, four tissue cores from four regions, two from the invasive margin (IM) and two from the center of tumor (CT), were randomly extracted from formalin-fixed paraffin-embedded (FFPE) tissue blocks in each case using a 1-mm-sized punch, and they were made into a tissue microarray (TMA). Pan-cytokeratin, vimentin, and CD3 were stained using an immunofluorescence assay for differential visualization of three primary cell types (carcinoma cells, stromal cells, and TILs, respectively) on the TMA slide. One geometric region of interest (ROI) was targeted in every TMA core without segregating cell types (total ROIs, n = 48). To measure mRNA expression, the target-specific oligonucleotide probes were counted using a NanoString nCounter instrument (NanoString Technologies). We used a fully validated Immune Pathways Panel (NanoString Technologies), covering 73 immuno-oncology-related genes, five housekeeping genes, and six isotype negative controls. The genes and their immune signature annotations are listed in Supplementary Table S2. Digital counts from oligonucleotide barcodes were first normalized to External RNA Control Consortium (ERCC) spike-in controls and then to negative controls32. The negative control barcodes were hybridized without tissue targets, normalization to which was adjusted for nonspecific nucleotide binding, and were used to establish signal-to-noise ratios for gene expression analysis.
Gene expression signatures of immune cell population and function
Normalized gene expression levels representing 22 major immune cell populations were obtained from a previous report (Leukocyte signature matrix 22 [LM22] or CIBERSORT) (Supplementary Table S3)33. Gene sets curated with immunologic functions were downloaded from the Immunology Database and Analysis Portal (IMMPORT) website (https://www.immport.org/shared/genelists) (Supplementary Table S4)34. Additional immunologically relevant functions were manually assigned according to the functional annotations provided in the Immune Pathways Panel (NanoString Technologies) and previous literature (Supplementary Table S5). The expression of matched genes in our data was used to compute immune cell types using the weight of relative expression in LM22. For IMMPORT and manual functional annotations, the expression levels of the included genes were averaged without weighting, except for BCL2, which was assumed to counteract apoptosis, opposite to FAS and TNF35.
Bioinformatics analysis
Differentially expressed genes (DEGs) were identified between the TIL subgroups (TIL-high vs. TIL-low subgroups; or TIL-low/heterogeneity-high vs. TIL-low/heterogeneity-low subgroups) using a nonparametric Mann–Whitney U test at a significance level of Benjamini–Hochberg false discovery rate (FDR) < 0.05 in R ver. 3.6.3 (The R Foundation, Vienna, Austria). Gene enrichment analysis was conducted for Gene Ontology-Biologic Processes (GOBPs)36,37, MSigDB Biocarta pathway38, and KEGG pathway39 using the ToppGene suite40. A formal Gene Set Enrichment Analysis (GSEA) was performed against custom gene sets retrieved from literature38. These included 28 CRC gene sets that were relevant to immune response, prognosis, and genetic features41 and 123 melanoma gene sets that were significantly (fold change ≥ 2.0 and FDR < 0.05) upregulated in the good response group compared with those in the poor response group to anti-PD-1 therapy (nivolumab or pembrolizumab) (Supplementary Table S6)42.
Statistics
Fisher’s exact test and Kruskal–Wallis test were used to compare categorical and continuous variables, respectively. Pearson’s coefficient was used to analyze the correlation between Shannon and Simpson indices. Kaplan–Meier plots and log-rank tests were used to compare DFS according to the density and spatial heterogeneity of TILs. Univariate and multivariate survival analyses were performed using the Cox proportional hazards regression model. The optimal values of high and low evenness indices were determined based on the maximal statistical significance (the lowest log-rank p value that was defined by maximally selected rank statistics) using the ClinicoPath package ver. 0.0.2 in Jamovi software ver. 1.6.23 (The jamovi project, Sydney, Australia). All statistical analyses were conducted using R ver. 3.6.3 (The R Foundation).
Results
Prognostic significance of the spatial heterogeneity of TILs in MSI-H CRCs
The CD3+ and CD8+ TILs were separately counted in every patch from the whole tumor area on a whole-slide image of each case using the digital pathology technique in 73 MSI-H CRCs (discovery cohort). In addition to the average density of CD3+ and CD8+ TILs, the spatial distribution heterogeneity of TIL densities was measured based on the Shannon and Simpson evenness indices (Fig. 1a). A tumor showing more spatial heterogeneity of TIL densities had lower evenness indices than that showing less, even though the average TIL densities of both tumors were the same (Fig. 1b). The average TIL density and Shannon and Simpson evenness indices computed in representative cases are presented in Fig. 1a. The Shannon and Simpson indices were closely inter-correlated (Pearson coefficient r = 0.943 for CD3+ TILs and 0.922 for CD8+ TILs, all p < 0.0001), indicating that both indices similarly reflect the spatial evenness of TIL densities (Fig. 1c).

a Heatmaps of patch-level densities of CD3+ TILs in representative cases of CD3+ TIL-high (CD3-H) (left), CD3+ TIL-low (CD3-L)/heterogeneity-low (Hetero-L) (middle), and CD3-L/heterogeneity-high (Hetero-H) (right) MSI-H CRCs with average TIL density and spatial TIL heterogeneity indices. b Schematic illustrations of MSI-H CRC cases showing the same average TIL densities but different TIL evenness indices. c Shannon and Simpson indices are significantly inter-correlated for both CD3+ and CD8+ TILs in MSI-H CRCs. d The CD3-L group is significantly associated with shorter DSF than that of the CD3-H group in MSI-H CRCs (discovery cohort, n = 73). e The CD8-L group is significantly associated with shorter DSF than that of the CD8-H group in MSI-H CRCs (discovery cohort, n = 73). f–i Spatial heterogeneity of TILs can more accurately stratify DFS subgroups in the TIL-low group of MSI-H CRCs (discovery cohort, n = 73). The combined parameters included CD3+ TIL density and Shannon index (f), CD3+ TIL density and Simpson index (g), CD8+ TIL density and Shannon index (h), and CD8+ TIL density and Simpson index (i).
TIL density is a well-known prognostic factor in CRCs1,4,5,6. To find the TIL density that optimally stratifies prognosis in MSI-H CRCs, the average densities of CD3+ and CD8+ TILs were first divided into quartiles. According to Kaplan–Meier survival analyses, the 1st (0–25%) and 2nd (25–50%) quartile subgroups showed the significant overlap of their DFS curves (Supplementary Fig. S1). A similar pattern was also observed between the 3rd (50–75%) and 4th (75–100%) quartile subgroups (Supplementary Fig. S1). These findings suggest that the 1st and 2nd quartile subgroups could be integrated into a low TIL density group (TIL-low; 0–50%), and the 3rd and 4th quartile subgroups could be integrated into a high TIL density group (TIL-high; 50–100%). Therefore, the median CD3+ or CD8+ TIL densities were used to classify TIL-high and TIL-low groups in MSI-H CRCs. As expected, the CD3+ TIL-low group (p = 0.00077) and CD8+ TIL-low group (p = 0.0045) were significantly associated with poor DFS in MSI-H CRCs (Fig. 1d, e).
Next, we examined if the spatial heterogeneity of TILs, when combined to establish TIL-low/heterogeneity-high and TIL-low/heterogeneity-low subgroups, had an additional impact on the survival differences of TIL-low tumors. Survival analyses using the statistically determined (at the lowest log-rank p value), optimal cutoff values of TIL spatial heterogeneity indices showed that the TIL-low/heterogeneity-high, TIL-low/heterogeneity-low, and TIL-high subgroups were significantly associated with poor, intermediate-to-good, and good prognoses, respectively, in all combinations, including CD3+ TILs with a Shannon index (p = 0.0017), CD3+ TILs with a Simpson index (p = 0.00039), CD8+ TILs with a Shannon index (p = 0.0012), and CD8+ TILs with a Simpson index (p < 0.0001) (Fig. 1f–i).
The CD3+ or CD8+ TIL-high group was not further divided according to TIL spatial heterogeneity status because the very low risk of death or tumor recurrence observed in the TIL-high group made it unnecessary to subdivide the TIL-high tumors. (Fig. 1d, e). Moreover, we subclassified the TIL-high groups based on TIL spatial heterogeneity and found that almost all TIL-high cases showed low spatial heterogeneity of TILs (Supplementary Fig. S2a). The only exception was that the CD3+ TIL-high group was subclassified by the Simpson evenness index into TIL-high/heterogeneity-low (n = 30) and TIL-high/heterogeneity-high (n = 7) subgroups in the discovery cohort (Supplementary Fig. S2a). However, survival analysis performed using this classification revealed no significant difference in DFS between the CD3+ TIL-high/heterogeneity-high and CD3+ TIL-high/heterogeneity-low subgroups (log-rank p = 0.69) (Supplementary Fig. S2b). These findings collectively support that the TIL spatial heterogeneity-based subclassification of the TIL-high group is insignificant and unnecessary in MSI-H CRCs.
To independently confirm these findings, we additionally collected 93 MSI-H CRCs (external validation cohort) and conducted survival analyses using TIL density and spatial heterogeneity parameters in the same way. The prognostic significance of TIL density and spatial heterogeneity in MSI-H CRCs was also confirmed in the validation cohort (Fig. 2). The CD3+ or CD8+ TIL-low group was significantly associated with worse DFS in MSI-H CRCs (Fig. 2a, b). The TIL-low/heterogeneity-high, TIL-low/heterogeneity-low, and TIL-high subgroups were significantly associated with poor, intermediate, and good DFS, respectively, in all four combinations (Fig. 2c–f).

a The CD3+ TIL-low (CD3-L) group is significantly associated with shorter DSF than that of the CD3+ TIL-high (CD3-H) group in MSI-H CRCs (validation cohort, n = 93). b The CD8+ TIL-low (CD8-L) group is significantly associated with shorter DSF than that of the CD8+ TIL-high (CD8-H) group in MSI-H CRCs (validation cohort, n = 93). c–f Spatial heterogeneity of TILs can more accurately stratify DFS subgroups in the TIL-low group of MSI-H CRCs (validation cohort, n = 93). The combined parameters included CD3+ TIL density and Shannon index (c), CD3+ TIL density and Simpson index (d), CD8+ TIL density and Shannon index (e), and CD8+ TIL density and Simpson index (f).
In addition, univariate Cox regression analysis identified that the TIL-low/heterogeneity-high subgroups were specific for poor DFS in the discovery and validation cohorts (all p < 0.01) (Table 1). Multivariate Cox models adjusted for TNM stage further confirmed that all the TIL-low/heterogeneity-high subgroups were independent poor prognostic factors in MSI-H CRCs in both cohorts (CD3+ TILs with a Shannon index (adjusted hazard ratio [HR] (95% confidence interval [CI]) = 9.82 (3.11–31.03), p < 0.001 in the discovery cohort and 5.92 (1.96–17.89), p = 0.002 in the validation cohort), CD3+ TILs with a Simpson index (adjusted HR (95% CI) = 10.81 (1.37–85.24), p = 0.024 in the discovery cohort and 5.92 (1.96–17.89), p = 0.002 in the validation cohort), CD8+ TILs with a Shannon index (adjusted HR (95% CI) = 8.37 (2.27–30.78), p = 0.001 in the discovery cohort and 5.21 (1.42–19.04), p = 0.013 in the validation cohort), and CD8+ TILs with a Simpson index (adjusted HR (95% CI) = 9.36 (2.12–41.42), p = 0.003 in the discovery cohort and 4.43 (1.38–14.21), p = 0.012 in the validation cohort) (Table 1).
These findings indicate that the quantitatively measured spatial heterogeneity of TILs can robustly classify the low TIL density group of MSI-H CRCs into further stratified prognostic subgroups.
Clinicopathologic associations of TIL density/spatial heterogeneity in MSI-H CRCs
We also investigated the clinicopathologic characteristics of the three TIL subgroups (TIL-low/heterogeneity-high, TIL-low/heterogeneity-low, and TIL-high subgroups) in the discovery cohort classified by CD3/Shannon index (Supplementary Table S7), CD3/Simpson index (Supplementary Table S8), CD8/Shannon index (Supplementary Table S9), and CD8/Simpson index (Supplementary Table S10) and those in the validation cohort classified by CD3/Shannon index or CD3/Simpson index (Supplementary Table S11), CD8/Shannon index (Supplementary Table S12), and CD8/Simpson index (Supplementary Table S13). Notably, the TIL-low/heterogeneity-high subgroups were consistently enriched with male patients with or without statistical significance (Supplementary Tables S7–S13). In the discovery cohort, although statistical significance was not reached (p = 0.054), the CD8+ TIL-low/Simpson heterogeneity-high subgroup was enriched with stage IV cases (Supplementary Table S10). In the validation cohort, the TIL-low/heterogeneity-high subgroups were significantly associated with advanced stage (III or IV) (p < 0.001 for CD3/Shannon, <0.001 for CD3/Simpson, 0.005 for CD8/Shannon, and 0.001 for CD8/Simpson) (Supplementary Tables S11–S13). In contrast to stage, tumor grade was not associated with the TIL density/spatial heterogeneity subgroups in MSI-H CRCs (Supplementary Tables S7–S13). In the discovery cohort, 38 (52.1%) were molecularly determined to be a hereditary subtype (Lynch or Lynch-like syndrome) (Supplementary Table S1), and this hereditary MSI-H subtype was more prevalent in the TIL-low/heterogeneity-high subgroups with no or marginal statistical significance (p = 0.13, 0.86, 0.09, and 0.048 for CD3/Shannon, CD3/Simpson, CD8/Shannon, and CD8/Simpson, respectively) (Supplementary Tables S7–S10).
Spatial profiling of immune-related expression signatures differed by TIL density in MSI-H CRCs
To identify spatially-resolved, differential immune gene expression signatures among the three TIL subgroups (TIL-low/heterogeneity-high, TIL-low/heterogeneity-low, and TIL-high subgroups) of MSI-H CRCs, GeoMx DSP analysis was conducted for the 48 ROIs that were collected from the IM and CT of 12 representative cases (Fig. 3a). Overall, four (33.3%) and eight (66.7%) cancers were CD3+ TIL-high and CD3+ TIL-low, respectively, while five (41.7%) and seven (58.3%) were CD8+ TIL-high and CD8+ TIL-low, respectively, and one (8.3%) was CD3+ TIL-low and CD8+ TIL-high. To identify immune-related gene expression profiles that differed by TIL density in MSI-H CRCs, we identified DEGs between the TIL-high and TIL-low groups (Mann–Whitney U test FDR < 0.05). The IM and CT regions were analyzed together because they showed similar gene expression levels in overall comparison between 24 IM and 24 CT ROIs (FDR > 0.05). As expected, TIL-high tumors showed overall upregulation of genes related to immune cells and their functions: 45 genes were upregulated in the CD3+ TIL-high group compared with the CD3+ TIL-low group (Fig. 3b) and 31 genes were upregulated in the CD8+ TIL-high group compared with the CD8+ TIL-low group (Fig. 3c). Furthermore, 29 upregulated genes were common in both comparisons, including CD3E, CD8A, IFNG, ARG1, B2M, CCL5, CXCL10, IDO1, HLA-DQA1, ICAM1, MKI67, and FAS (Fig. 3d). These commonly upregulated genes were significantly (FDR < 0.05) enriched in immune-related GOBPs (Fig. 3e) and pathways (Fig. 3f), including T cells, cytokines, interferon-γ, and antigen processing and presentation. To assess differences in the global immune contexture between CD3+ TIL-high/CD8+ TIL-high (TIL-high) and CD3+ TIL-low/CD8+ TIL-low (TIL-low) groups, we deconvoluted the DSP-based gene expression data to identify immune cell types and functions. Nearly all immune cell components, including T, B, NK, dendritic, mast, and plasma cells, monocytes, macrophages, eosinophils, and neutrophils (all, p < 0.0001), were significantly higher in the TIL-high group than those in the TIL-low group (Fig. 3g). Likewise, immunologic functions and pathways related to the T cell receptor pathway (p = 0.0004), B cell receptor pathway (p = 0.0047), antigen presentation (p < 0.0001), antimicrobials (p < 0.0001), chemokines (p < 0.0001), cytokines (p = 0.0001), cytokine receptors (p = 0.0046), natural killer cells (p < 0.0001), interferons (p = 0.0018), interferon receptors (p = 0.009), myeloid cells (p < 0.0001), adhesion/migration (p = 0.0005), proliferation (p < 0.0001), and apoptosis (p = 0.0061) were also significantly upregulated in the TIL-high group compared with those in the TIL-low group (Fig. 3g). In contrast, only IL6 and IL12B were significantly upregulated in both CD3+ TIL-low and CD8+ TIL-low groups (Fig. 3b, c), consistent with the modest upregulation of interleukin signatures in TIL-low tumors (Fig. 3g). In addition, although the immune signatures were similar across different regions in the TIL-high group, the TIL-low group showed intratumoral or intertumoral heterogeneity (Fig. 3g).

a Four ROIs selected from two IM and two CT areas per MSI-H CRC tissue sample were subjected to GeoMx DSP analysis using an immuno-oncology gene panel. Cancer cells, stroma, and T cells are stained with pan-cytokeratin (green), vimentin (yellow), and CD3 (red), respectively. b DEGs identified from comparison between CD3+ TIL-high (CD3-H) and CD3+ TIL-low (CD3-L) groups of MSI-H CRCs (false discovery rate [FDR] < 0.05). c DEGs identified from comparison between CD8+ TIL-high (CD8-H) and CD8+ TIL-low (CD8-L) groups of MSI-H CRCs (FDR < 0.05). d The 29 commonly upregulated genes identified from DEGs of CD3-H (45 genes) and CD8-H (31 genes) groups of MSI-H CRCs (“TIL-H common upregulated genes”). e Gene Ontology-Biologic Processes (GOBPs) significantly enriched in the TIL-H common upregulated genes. f Gene signaling pathways significantly enriched in the TIL-H common upregulated genes. g Immune cell population and immunologic signatures curated based on LM22, IMMPORT, and manual gene sets. Mann–Whitney comparisons between the CD3+ TIL-high/CD8+ TIL-high (TIL-H) and CD3+ TIL-low/CD8+ TIL-low (TIL-L) groups of MSI-H CRCs were performed at the *p < 0.05, **p < 0.01, or ***p < 0.001 significance level. Low and high spatial heterogeneity groups (Hetero-L and Hetero-H, respectively) determined by both Shannon and Simpson indices are presented at the bottom of the heatmap.
Spatial profiling of immune-related expression signatures differed by TIL spatial heterogeneity in MSI-H CRCs
As the next step of DSP analysis, to unravel immune-related gene expression features that differed by TIL spatial heterogeneity in MSI-H CRCs, we compared the gene expression data between the TIL-low/heterogeneity-high and TIL-low/heterogeneity-low subgroups (Fig. 4). Genes related to immune checkpoints [CD274 (gene encoding PD-L1), VSIR (gene encoding VISTA), CTLA4, ICOSLG, and CD86], chemokine and cytokine receptors (CXCR6 and CMKLR1), and myeloid cells (IDO1 and ARG1) were significantly upregulated in the TIL-low/heterogeneity-high subgroup compared with those in the TIL-low/heterogeneity-low subgroup (Fig. 4a). In contrast, there were no upregulated DEGs in the TIL-low/heterogeneity-low subgroup. GOBP analysis showed that the upregulated genes in the TIL-low/heterogeneity-high subgroup were enriched in pathways related to lymphocyte and leukocyte activity, immune regulatory functions, cytokines, cell adhesion, cell development, cell death, and response to stimuli (Fig. 4b). Notably, consistent with the TIL spatial heterogeneity profiles, intratumoral interregional differences in the immune expression signatures were more variable in the TIL-low/heterogeneity-high subgroup than those in the TIL-low/heterogeneity-low subgroup (Figs. 3g, 4c). Similar to the DEG pathway data, deconvolutional analysis for profiling immune cell types and functional signatures also revealed that immune components in the TIL-low/heterogeneity-high subgroup were particularly enriched in immune checkpoints (p = 0.0006), chemokine receptors (p = 0.0006), and myeloid cells (p = 0.0011) (Fig. 4c). Immune checkpoint molecules, chemokine receptors, and myeloid cells have been recognized to affect response to ICI therapy43,44. Furthermore, the immune regulation pathways enriched in the TIL-low/heterogeneity-high subgroup could affect immunomodulation functions, which are possibly associated with response to ICIs. Therefore, we further conducted GSEA against two immuno-oncology-related gene sets retrieved from previous reports (Supplementary Table S4): one was an immune response- and survival-related gene set in CRCs, and the other was a gene set related to good response to anti-PD-1 immunotherapy in melanoma41,42. The TIL-low/heterogeneity-high subgroup was significantly enriched with these two gene sets (Fig. 4d), suggesting that the TIL-low/heterogeneity-high subgroup of MSI-H CRCs may be potentially beneficial to ICI treatment, although the TIL-low nature of the tumors is generally regarded to be disadvantageous to immunotherapy. The above findings collectively suggest that the spatial heterogeneity of TILs can determine not only prognostically but also immunologically distinct sub-entities within TIL-low MSI-H CRCs.

a Compared with the TIL-low/heterogeneity-low (TIL-L/Hetero-L) subgroup, 22 genes were significantly upregulated in the TIL-low/heterogeneity-high (TIL-L/Hetero-H) subgroup of MSI-H CRCs, which included immune checkpoints, cytokines, and myeloid cells-related genes. b The number of Gene Ontology-Biologic Process (GOBP) terms enriched for the upregulated genes in the TIL-L/Hetero-H tumors. c Immune cell population and immunologic signatures curated based on LM22, IMMPORT, and manual gene sets. Mann–Whitney comparisons between the TIL-L/Hetero-L and TIL-L/Hetero-H subgroups of MSI-H CRCs are performed at the *p < 0.05, **p < 0.01, or ***p < 0.001 significance level. d Gene sets related to the immune response in CRCs (left) and favorable response to PD-1 blockade (right) were positively correlated with the TIL-L/Hetero-H subgroup of MSI-H CRCs.
Discussion
Although previous studies on the clinical and prognostic impacts of TIL density in cancers are more dominant, several previous studies have investigated the implications of the spatial distribution of TILs. For example, Masugi et al. reported that the spatial distribution pattern of CD8+ TILs can refine the prognostic value of TILs in pancreatic cancers45. In detail, CD8+ TILs in the CT area, but not those in the IM area, were significantly associated with patient survival in pancreatic cancers. In fact, previous studies exploring the spatial context of TILs frequently focused on the region-specific significance of TILs, similar to the aforementioned study by Masugi and colleagues. For example, König et al. compared the associations of treatment responses between TILs in the CT and IM areas in breast cancers46. Although recent investigations also revealed the prognostic significance of the spatial architecture of TILs in lung or gynecologic cancers using computational analysis of whole-slide images with or without the aid of artificial intelligence47,48, our present study focused on applying a simple, easily interpretable index (Shannon or Simpson evenness index) for quantifying TIL spatial heterogeneity. We successfully proved that these indices could be used as potential prognostic markers in MSI-H CRCs when combined with average TIL density. Collectively, compared with previous studies by other researchers, our study concentrated more on the value of TIL spatial heterogeneity itself and further provided novel data regarding the differential spatial profiles of immune-related gene expression signatures according to the spatial TIL subtypes in MSI-H CRCs. Clinical implications of our study results are graphically summarized in Fig. 5.

Schematic summary of this study.
Based on the spatial distribution of TILs, tumor immune phenotypes have been conceptually classified as two or three subtypes49,50. Tumor immune phenotypes are broadly dichotomized into immune-hot and -cold types. Immune-hot tumors are characterized by densely infiltrated, intratumorally-distributed TILs that are functionally inhibited by immune checkpoints and are expected to respond well to ICI therapy. In contrast, immune-cold tumors show a lack of intratumoral infiltration of TILs and are generally associated with poor response to ICI therapy. Immune-cold tumors can be further divided into immune-desert and -excluded types. The immune-desert phenotype is characterized by the near absence of both intratumoral and peritumoral TILs, whereas in the immune-excluded phenotype, TILs do not efficiently infiltrate into the intratumoral area rather than being absolutely deficient. Based on our finding that the TIL-low/heterogeneity-high subgroup represents tumors in which TILs unevenly and inefficiently infiltrate, these tumors can be conceptually matched with the immune-excluded phenotype. On the other hand, the TIL-low/heterogeneity-low tumors are characterized by widely scanty infiltration of TILs and thus, are similar to the immune-desert phenotype. Although the consensus criteria for classifying immune-excluded and -desert phenotypes in tumor samples have not yet been established, our approach to quantitatively evaluating the spatial heterogeneity of TILs can provide a simple and useful method for practically classifying immune-excluded and -desert phenotypes in surgically-resected tumor tissues (Fig. 5).
There are several limitations to our present study. First, our study cohort was confined to CRCs, especially the MSI-H molecular subtype. Although the MSI-H subtype comprises a limited portion of overall CRCs, we intensively collected and tested MSI-H CRCs in this study because there were several advantages of MSI-H CRCs in investigating the implications of TIME. MSI-H CRCs generally display greater intertumoral variations in TIME features, including the density of TILs, compared with those in microsatellite stable (MSS) CRCs12. In our study cohort, these larger variations in immune parameters in MSI-H CRCs could allow various compositions of the three tumor immune phenotypes, including immune-hot, -excluded, and -desert types. MSS CRCs were determined to be less suitable for our exploration because most MSS CRCs are known to be immune-desert phenotypes and were thus excluded from our current study. However, it is still necessary to validate our findings in larger CRC cohorts encompassing MSS CRCs. In addition, our main finding, the prognostic and immunologic significance of the quantitatively measured spatial heterogeneity of TILs, should be further confirmed in other solid tumors besides CRCs. The second limitation of our study was the lack of real-world data to demonstrate the therapeutic relevance of our findings. Although TIL-low/heterogeneity-high MSI-H CRCs may be likely to respond well to ICI therapy based on the observation that gene sets related to immunotherapy response were enriched in the TIL-low/heterogeneity-high subgroup (Fig. 4d), there were only three cases that were treated with ICI in our retrospective MSI-H CRC cohort. Therefore, it was practically impossible to evaluate the relevance of TIL spatial heterogeneity to immunotherapeutic responses in our samples. Further clinical studies or trials will be necessary to assess the relationship between TIL spatial heterogeneity subtypes and immunotherapy responses in various tumors.
In conclusion, the spatial heterogeneity of TILs can be considered a significant factor for precisely stratifying prognostic immune subgroups of MSI-H CRCs. The TIL-low/heterogeneity-high subgroup conceptually represents the immune-excluded phenotype and may be associated with a relatively good response to ICI-based immunotherapy, although the subgroup showed the worst prognosis in MSI-H CRCs. The potential value of the quantitatively measured spatial heterogeneity of TILs as a prognostic and/or predictive biomarker should be further evaluated in various malignancies through future clinical studies.
Data availability
The raw dataset generated from GeoMx DSP analysis during the current study are available in the Mendeley data repository (https://doi.org/10.17632/jcyt9grfjv.1). The other datasets generated or analyzed in the study are available from the corresponding author upon reasonable request.
References
Bruni D, Angell HK & Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat Rev Cancer 20, 662–680 (2020).
Fridman WH, Zitvogel L, Sautes-Fridman C & Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol 14, 717–734 (2017).
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313, 1960–1964 (2006).
Idos GE, Kwok J, Bonthala N, Kysh L, Gruber SB & Qu C. The Prognostic Implications of Tumor Infiltrating Lymphocytes in Colorectal Cancer: A Systematic Review and Meta-Analysis. Sci Rep 10, 3360 (2020).
Lanzi A, Pages F, Lagorce-Pages C & Galon J. The consensus immunoscore: toward a new classification of colorectal cancer. Oncoimmunology 9, 1789032 (2020).
Bae JM, Yoo SY, Kim JH & Kang GH. Immune landscape and biomarkers for immuno-oncology in colorectal cancers. J Pathol Transl Med 54, 351–360 (2020).
Vilar E & Gruber SB. Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Oncol 7, 153–162 (2010).
Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov 5, 43–51 (2015).
Makaremi S, Asadzadeh Z, Hemmat N, Baghbanzadeh A, Sgambato A, Ghorbaninezhad F et al. Immune Checkpoint Inhibitors in Colorectal Cancer: Challenges and Future Prospects. Biomedicines 9, 1075 (2021).
Giannini R, Zucchelli G, Giordano M, Ugolini C, Moretto R, Ambryszewska K et al. Immune Profiling of Deficient Mismatch Repair Colorectal Cancer Tumor Microenvironment Reveals Different Levels of Immune System Activation. J Mol Diagn 22, 685–698 (2020).
Yoon HH, Shi Q, Heying EN, Muranyi A, Bredno J, Ough F et al. Intertumoral Heterogeneity of CD3(+) and CD8(+) T-Cell Densities in the Microenvironment of DNA Mismatch-Repair-Deficient Colon Cancers: Implications for Prognosis. Clin Cancer Res 25, 125–133 (2019).
Kim JH, Seo MK, Lee JA, Yoo SY, Oh HJ, Kang H et al. Genomic and transcriptomic characterization of heterogeneous immune subgroups of microsatellite instability-high colorectal cancers. J Immunother Cancer 9, e003414 (2021).
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 372, 2509–2520 (2015).
Andre T, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C et al. Pembrolizumab in microsatellite-instability-high advanced colorectal cancer. N Engl J Med 383, 2207–2218 (2020).
Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol 18, 1182–1191 (2017).
Le DT, Kim TW, Van Cutsem E, Geva R, Jager D, Hara H et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J Clin Oncol 38, 11–19 (2020).
Lee JA, Yoo SY, Oh HJ, Jeong S, Cho NY, Kang GH et al. Differential immune microenvironmental features of microsatellite-unstable colorectal cancers according to Fusobacterium nucleatum status. Cancer Immunol Immunother 70, 47–59 (2021).
Kim JH, Hong JH, Choi YL, Lee JA, Seo MK, Lee MS et al. NTRK oncogenic fusions are exclusively associated with the serrated neoplasia pathway in the colorectum and begin to occur in sessile serrated lesions. J Pathol 255, 399–411 (2021).
Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 58, 5248–5257 (1998).
Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD et al. QuPath: Open source software for digital pathology image analysis. Sci Rep 7, 16878 (2017).
Loughrey MB, Bankhead P, Coleman HG, Hagan RS, Craig S, McCorry AMB et al. Validation of the systematic scoring of immunohistochemically stained tumour tissue microarrays using QuPath digital image analysis. Histopathology 73, 327–338 (2018).
Simpson E. Measurement of diversity. Nature 163, 688 (1949).
Shannon CE. A mathematical theory of communication. The Bell System Technical Journal 27, 379–423 (1948).
Morris EK, Caruso T, Buscot F, Fischer M, Hancock C, Maier TS et al. Choosing and using diversity indices: insights for ecological applications from the German Biodiversity Exploratories. Ecol Evol 4, 3514–3524 (2014).
Evans JA, Carlotti E, Lin ML, Hackett RJ, Haughey MJ, Passman AM et al. Clonal transitions and phenotypic evolution in Barrett’s esophagus. Gastroenterology 162, 1197–1209 e1113 (2022).
Nelson LS, Mansfield JR, Lloyd R, Oguejiofor K, Salih Z, Menasce LP et al. Automated prognostic pattern detection shows favourable diffuse pattern of FOXP3(+) Tregs in follicular lymphoma. Br J Cancer 113, 1197–1205 (2015).
Levy-Jurgenson A, Tekpli X, Kristensen VN & Yakhini Z. Spatial transcriptomics inferred from pathology whole-slide images links tumor heterogeneity to survival in breast and lung cancer. Sci Rep 10, 18802 (2020).
Andersson A, Larsson L, Stenbeck L, Salmen F, Ehinger A, Wu SZ et al. Spatial deconvolution of HER2-positive breast cancer delineates tumor-associated cell type interactions. Nat Commun 12, 6012 (2021).
Campisciano G, Florian F, D’Eustacchio A, Stankovic D, Ricci G, De Seta F et al. Subclinical alteration of the cervical-vaginal microbiome in women with idiopathic infertility. J Cell Physiol 232, 1681–1688 (2017).
Nagendra H. Opposite trends in response for the Shannon and Simpson indices of landscape diversity. Applied Geography 22, 175–186 (2002).
Pielou EC. The measurement of diversity in different types of biological collections. Journal of Theoretical Biology 13, 131–144 (1966).
Merritt CR, Ong GT, Church SE, Barker K, Danaher P, Geiss G et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat Biotechnol 38, 586–599 (2020).
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12, 453–457 (2015).
Bhattacharya S, Dunn P, Thomas CG, Smith B, Schaefer H, Chen J et al. ImmPort, toward repurposing of open access immunological assay data for translational and clinical research. Sci Data 5, 180015 (2018).
Yin XM. Signal transduction mediated by Bid, a pro-death Bcl-2 family proteins, connects the death receptor and mitochondria apoptosis pathways. Cell Res 10, 161–167 (2000).
Gene Ontology Consortium. The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res 49, D325–D334 (2021).
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29 (2000).
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550 (2005).
Kanehisa M & Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30 (2000).
Chen J, Bardes EE, Aronow BJ & Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 37, W305-W311 (2009).
Lal N, Beggs AD, Willcox BE & Middleton GW. An immunogenomic stratification of colorectal cancer: Implications for development of targeted immunotherapy. Oncoimmunology 4, e976052 (2015).
Gide TN, Quek C, Menzies AM, Tasker AT, Shang P, Holst J et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 35, 238–255 e236 (2019).
Mollica Poeta V, Massara M, Capucetti A & Bonecchi R. Chemokines and chemokine receptors: new targets for cancer immunotherapy. Front Immunol 10, 379 (2019).
Peranzoni E, Ingangi V, Masetto E, Pinton L & Marigo I. Myeloid cells as clinical biomarkers for immune checkpoint blockade. Front Immunol 11, 1590 (2020).
Masugi Y, Abe T, Ueno A, Fujii-Nishimura Y, Ojima H, Endo Y et al. Characterization of spatial distribution of tumor-infiltrating CD8(+) T cells refines their prognostic utility for pancreatic cancer survival. Mod Pathol 32, 1495–1507 (2019).
Konig L, Mairinger FD, Hoffmann O, Bittner AK, Schmid KW, Kimmig R et al. Dissimilar patterns of tumor-infiltrating immune cells at the invasive tumor front and tumor center are associated with response to neoadjuvant chemotherapy in primary breast cancer. Bmc Cancer 19, 120 (2019).
Park S, Ock CY, Kim H, Pereira S, Park S, Ma M et al. Artificial Intelligence-Powered Spatial Analysis of Tumor-Infiltrating Lymphocytes as Complementary Biomarker for Immune Checkpoint Inhibition in Non-Small-Cell Lung Cancer. J Clin Oncol 40, 1916–1928 (2022).
Azarianpour S, Corredor G, Bera K, Leo P, Fu P, Toro P et al. Computational image features of immune architecture is associated with clinical benefit and survival in gynecological cancers across treatment modalities. J Immunother Cancer 10 (2022).
Chen DS & Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature 541, 321–330 (2017).
Liu YT & Sun ZJ. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics 11, 5365–5386 (2021).
Acknowledgements
We thank Seoul National University Hospital Medical Research Collaborating Center for partially supporting statistical analysis in the study.
Funding
This study was supported by a grant from the SNUH Research Fund (04-2020-0550; to JHK), a faculty research grant of Yonsei University College of Medicine (6-2021-0144; to MJ), a grant from Seoul National University College of Medicine (800-20210387; to JHK), a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute funded by the Korea government (Ministry of Health and Welfare) (HI21C0977; to MJ), and the National Research Foundation of Korea grants funded by the Korea government (Ministry of Science and ICT) (NRF-2016R1C1B2010627; NRF-2019R1F1A1059535; to JHK).
Author information
Authors and Affiliations
Contributions
MJ and JHK performed study concept and design. MJ, JAL, S-YY, JMB, GHK, and JHK collected tissue samples and clinicopathologic data. MJ, JAL, S-YY, and JHK provided acquisition, analysis, and interpretation of data. MJ and JHK conducted statistical analyses. MJ and JHK wrote and revised the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was conducted in compliance with the ethical guidelines of the 2013 Declaration of Helsinki and was approved by the Institutional Review Boards of Seoul National University Hospital (IRB No. 1804-036-935; and 1805-018-944) and Severance Hospital (IRB No. 4-2021-1130). The study samples collected from Seoul National University Hospital were previously registered in the Cancer Tissue Bank of Seoul National University Hospital with informed consent obtained from all patients about the research use of their tissues. The study samples collected from Severance Hospital were exempted from informed consent acquisition from patients by the Institutional Review Board because the study used only retrospective, anonymized tissue samples.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Jung, M., Lee, J.A., Yoo, SY. et al. Intratumoral spatial heterogeneity of tumor-infiltrating lymphocytes is a significant factor for precisely stratifying prognostic immune subgroups of microsatellite instability-high colorectal carcinomas. Mod Pathol 35, 2011–2022 (2022). https://doi.org/10.1038/s41379-022-01137-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-022-01137-0
This article is cited by
-
Clinical and biomarker results from a phase II trial of combined cabozantinib and durvalumab in patients with chemotherapy-refractory colorectal cancer (CRC): CAMILLA CRC cohort
Nature Communications (2024)
-
Single-cell morphological and topological atlas reveals the ecosystem diversity of human breast cancer
Nature Communications (2023)
-
Innovative molecular subtypes of multiple signaling pathways in colon cancer and validation of FMOD as a prognostic-related marker
Journal of Cancer Research and Clinical Oncology (2023)
