
Abstract
Though uncommon in melanoma, gene fusions may have therapeutic implications. Next generation sequencing-based clinical assays, designed to detect relevant gene fusions, mutations, and copy number changes, were performed on 750 melanomas (375 primary and 375 metastases) at our institution from 2014–2021. These included 599 (80%) cutaneous, 38 (5%) acral, 11 (1.5%) anorectal, 23 (3%) sinonasal, 27 (3.6%) eye (uveal/ conjunctiva), 11 (1.5%) genital (vulva/penile), and 41 (5.5%) melanomas of unknown primary. Sixteen fusions (2%) were detected in samples from 16 patients: 12/599 (2%) cutaneous, 2/38 (5%) acral, 1/9 (11%) vulva, 1/23(4.3%) sinonasal; and 12/16 (75%) fusions were potentially targetable. We identified two novel rearrangements: NAGS::MAST2 and NOTCH1::GNB1; and two fusions that have been reported in other malignancies but not in melanoma: CANT1::ETV4 (prostate cancer) and CCDC6::RET (thyroid cancer). Additional fusions, previously reported in melanoma, included: EML4::ALK, MLPH::ALK, AGAP3::BRAF, AGK::BRAF, CDH3::BRAF, CCT8::BRAF, DIP2B::BRAF, EFNB1::RAF1, LRCH3::RAF1, MAP4::RAF1, RUFY1::RAF1, and ADCY2::TERT. Fusion positive melanomas harbored recurrent alterations in TERT and CDKN2A, among others. Gene fusions were exceedingly rare (0.2%) in BRAF/RAS/NF1-mutant tumors and were detected in 5.6% of triple wild-type melanomas. Interestingly, gene rearrangements were significantly enriched within the subset of triple wild-type melanomas that harbor TERT promoter mutations (18% versus 2%, p < 0.0001). Thirteen (81%) patients were treated with immunotherapy for metastatic disease or in the adjuvant setting. Six of 12 (50%) patients with potentially actionable fusions progressed on immunotherapy, and 3/6 (50%) were treated with targeted agents (ALK and MEK inhibitors), 2 off-label and 1 as part of a clinical trial. One patient with an AGAP3::BRAF fusion positive melanoma experienced a 30-month long response to trametinib. We show that, detecting fusions, especially in triple wild-type melanomas with TERT promoter mutations, may have a clinically significant impact in patients with advanced disease who have failed front-line immunotherapy.
Similar content being viewed by others

Introduction
Melanoma accounts for the majority of deaths from skin cancers (9000 deaths per year) and its worldwide incidence is increasing1,2. Melanoma is often associated with cumulative solar damage from ultraviolet (UV) radiation, but it can occur at non-sun exposed anatomic locations, including mucosal, acral and uveal sites1. Treatment for melanoma ranges from surgical excision in cases of localized disease, to radiotherapy, chemotherapy, immunotherapy and targeted therapy in more complex or advanced cases3. Of special interest, are targeted agents that inhibit key pathways associated with melanoma carcinogenesis, such as the mitogen-activated protein kinase (MAPK) pathway, which involves the KIT, NRAS, BRAF and MEK1/2 genes4. BRAF inhibitors such as Vemurafenib and Dabrafenib as well as MEK inhibitors such as Trametinib and Cobimetinib have been shown to improve survival outcomes3. Other kinase inhibitors such as Imatinib, Sunitinib, Dasatinib and Nilotinib may also have activity in patients with melanoma harboring KIT mutations3.
The Tumor Cancer Genome Atlas (TCGA) has recognized four distinct subtypes of melanoma based on the molecular alteration present: mutant BRAF, mutant RAS (including NRAS, KRAS and HRAS genes), mutant NF1 and a triple wild-type subtype (lacking hotspot mutations in BRAF, NRAS, KRAS, HRAS and NF1)5. NF1 encodes a negative regulator of the RAS/MAPK pathway. Tumor suppressors TP53, CDKN2A and PTEN are often inactivated in all melanoma subtypes, by loss of function mutations or by whole gene deletion. Also frequently observed in melanomas, are TERT promoter mutations, which lead to increased expression of the TERT gene and to improved chromosomal stability in rapidly dividing cancer cells6,7. Ultraviolet radiation mutational signatures, resulting from cumulative solar damage and characterized by recurrent C to T transitions at the 3’ end of pyrimidine dimers, are commonly identified in cutaneous melanomas and associated with TERT promoter mutations8. While there is some overlap between the genetic signatures of cutaneous and mucosal melanomas, uveal melanomas are often characterized by mutations in GNAQ/GNA11, BAP1, SF3B1, and EIF1AX4,9.
Additionally, high-throughput, deep-sequencing technologies (next-generation sequencing) have allowed the identification of previously unknown alterations like gene fusions in a number of tumor types including sarcomas, carcinomas, gliomas and melanomas10,11. Some of these gene fusions are thought to function as driver mutations and the presence or absence of a gene fusion may enable risk stratification. Importantly, kinase fusions including genes such as ALK, ROS, RET, NTRK and FGFR family members have been identified in a variety of tumor types and can be targeted by available drugs11.
Although gene fusions are rare in melanoma, rearrangements involving the BRAF, RAF1, ALK, ROS1, NTRK1/2/3, MAP3K3, MAP3K8, PRKCA, and TRIM11 genes have been reported11,12,13,14,15,16. Interestingly, melanomas that contain kinase fusions have been reported to lack common driver mutations (BRAF, NRAS, NF1), further supporting the theory that gene rearrangements in melanoma represent driver mutations14. Identification of these driver mutations by next-generation sequencing may be clinically relevant if targeted therapy employed in other tumor types (e.g. ALK inhibitors in lung cancer) can be utilized in the treatment of melanoma17. In fact, in recent years, there have been several reports showing encouraging responses to specific inhibitors, in patients with fusion-associated melanomas18,19,20,21,22.
Of note, gene fusions have been associated with distinct subtypes, morphologic patterns and particular anatomic sites. Kinase fusions occur in Spitz neoplasms and BRAF fusions are frequently documented in Spitz tumors16,23. Spitz tumors characteristically affect children and adolescents. Histopathologically they exhibit a dome-shaped or wedged-shaped proliferation associated with epidermal hyperplasia and junctional retraction artifact and are comprised of spindle tumor cells with abundant eosinophilic cytoplasm. While most Spitz tumors are benign, rare Spitz melanomas are asymmetric, typically lack dermal maturation, and have frequent mitotic figures, spindle morphology and severe cytologic atypia. In contrast, conventional melanomas exhibit an epithelioid morphology, i.e. the tumor cells are polygonal with abundant cytoplasm24. BRAF fusion-positive Spitz tumors have been reported to have epithelioid, high-grade morphology and ALK fusions have been detected in up to 13% of acral lentiginous melanomas25,26. These findings suggest gene fusions may play a role in tumorigenesis, histomorphologic patterns and may be site-specific.
In this study we report how our institution’s approach of using next generation sequencing (NGS)-based assays for the clinical management of advanced cancer patients, resulted in the identification of rare and novel gene fusions in primary and metastatic melanomas from several anatomic sites (including cutaneous, anorectal, sinonasal, eye, genital and unknown site). Gene fusions were significantly enriched in triple wild-type melanomas harboring TERT promoter mutations (18%) and prompted the use of targeted therapies in 3 patients who progressed on immunotherapy, one of whom experienced a durable response to trametinib.
Materials and methods
Our study included 750 melanomas (375 primary and 375 metastases) submitted for clinical genetic profiling using targeted gene rearrangement testing and mutational analysis, from May 2014 until May 2021. Clinical parameters such as age, gender, treatment modality, and follow-up of the 16 patients whose tumors harbored fusions were collected from the patients’ electronic medical records. The available histologic sections of fusion-positive tumors were reviewed to evaluate the histologic patterns including predominant cell morphology.
Molecular analyses
Tumor genotyping was performed using two types of clinically validated Anchored Multiplex PCR (AMP)-based NGS assays27. Gene fusions were identified using ribonucleic acid (RNA)-based assays, designed to detect fusion transcripts involving genes commonly rearranged in solid tumors (Supplementary Table 1). Briefly, total nucleic acid extracted from formalin-fixed paraffin-embedded (FFPE) tumors (after histological review for tumor enrichment), was reverse transcribed with random hexamers, and processed to create double stranded complimentary deoxyribonucleic acid (cDNA). Two hemi-nested polymerase chain reactions (PCR) were performed to create a fully functional sequencing library, using custom designed FusionPlex Solid Tumor kit primers (ArcherDx Inc., Boulder, CO, USA). Illumina NextSeq 2 × 150 base paired-end sequencing results were aligned to the hg19 human genome reference using bwa-mem28. A laboratory-developed algorithm was used for fusion transcript detection and annotation.
Clinically validated AMP-based assays (SNAPSHOT-NGS, V1 and V2) were used to detect single nucleotide variants (SNV), small insertion/deletions (indel), and copy number variants in genomic DNA (Supplementary Table 1). Genomic DNA extracted from FFPE tumor tissue was enzymatically sheared, end-repaired, adenylated, and ligated with a half-functional adapter. A sequencing library targeting hotspots and exons in 99 cancer genes was generated using two hemi-nested polymerase chain reactions and custom designed VariantPlex kit primers (ArcherDx Inc., Boulder, CO, USA). Illumina MiSeq 2 × 151 base paired-end sequencing results were aligned to the hg19 human genome reference using BWA-MEM (Li 2009). MuTect and a laboratory-developed insertion/deletion analysis algorithm were used for SNV and indel variant detection, respectively29. Tumor mutational burden (TMB) is reported as a total absolute count of somatic mutations (including intronic, synonymous and non-synonymous variants) across the covered regions of the SNAPSHOT-NGS-V2 assay (Supplementary Table 1). Upon clinical validation using orthogonal assays, an absolute TMB count greater than or equal to 15 was established as the reference range for TMB-High on this assay. The TMB scores reported in this study are specific to our SNAPSHOT-NGS-V2 NGS panel and direct conversion of the reported absolute TMB count to other assays or standards is not possible.
Fluorescence in-situ hybridization
Interphase dual color break apart fluorescence in-situ hybridization (FISH) was performed on select cases to confirm NGS Solid Fusion Assay results, as previously detailed30. ALK gene rearrangements were analyzed using the Vysis ALK break-apart probe FISH Probe Kit (Abbot Molecular, Des Plaines, IL). The NOTCH1 rearrangement was confirmed using a commercially available NOTCH1 break apart FISH probe (Empire Genomics, Williamsville, NY). BRAF gene rearrangements were evaluated using homebrew FISH probes, consisting of two BAC clones flanking the BRAF gene: RP11-715H9 (BRAF 5’, Spectrum Green) and RP11-248P7 (BRAF 3’, Spectrum Orange). Briefly, 5-micron sections of FFPE tumor material were baked, deparaffinized, and hybridized with FISH probes in a Hybrite slide processor (Abbott Molecular Inc., Abbott Park, IL, USA), according to the manufacturer’s recommendations. After hybridization, the slides were washed, and the probes were visualized using an Olympus Bx61 microscope. FISH signals were evaluated in at least 50 nuclei in tumor-enriched areas, marked before slide processing, and the analysis was performed using the Cytovision software (Leica Biosystems, Buffalo Grove, IL, USA). A fused green-orange signal represents a normal gene. Fusion-positive nuclei show splitting of the green and orange probe signals further than two probes apart, in addition to the normal un-split green-orange signal. Samples were considered positive for a gene rearrangement, if more than 15% of scored tumor cells had split 5′ and 3′ FISH probe signals.
Immunohistochemistry and immunofluorescence
Immunohistochemical studies were performed on 5-micrometer-thick tissue sections using a Bond 3 automated immunostainer (Leica Microsystems, Bannockburn, IL, USA), with primary antibodies against ALK (predilute, 5 A4, Leica Microsystems) and Notch1 (D1E11, Cell Signaling Technology, Danvers, MA). Immunofluorescence studies were performed manually on 5-micrometer-thick tissue sections using standard protocol with primary antibodies against MAST2 (1:100, HPA039722, Sigma Aldrich, St. Louis, MA) and GNB1 (1:200, SAB2701168, Sigma Aldrich) and secondary antibodies (anti rabbit, Alexa Fluor 647 (cy5), 1:300, Thermo Fisher Scientific, Waltham, MA for MAST2; Goat anti rabbit, Alexa FluorTM Plus 594, A32740, Thermo Fisher Scientific for GNB1).
Results
Molecular analyses
Sixteen gene fusions were detected in 6 primary tumors and in 12 metastases of 16 patients (Fig. 1, Table 1). The presumed mechanisms underlying these fusions include translocation (7 cases), inversion (7 cases), and deletion (2 cases) (Table 1). Using public databases (COSMIC and TCGA) and published literature (PubMed) we identified 4 novel fusions, including 2 that have been reported in other malignancies but not in melanomas: CANT1::ETV4 (prostate cancer), CCDC6::RET (thyroid cancer), and 2 fusions that, to our knowledge, have not been previously described: NAGS::MAST2 and NOTCH1::GNB1 (Table 1, Fig. 2). The NOTCH1 rearrangement juxtaposes NOTCH1 exon 2 to the 5′-UTR of GNB1, immediately upstream of the GNB1 starting codon, and it does not appear to generate an in-frame chimeric protein between NOTCH1 and GNB1. However, it may function by inactivating NOTCH1, and/or by driving GNB1 expression under the control of the NOTCH1 promoter.

There was no evidence of loss of heterozygosity (LOH) for genes affected by nonsense or frameshift mutations.

The NAGS::MAST2 fusion has not been previously reported in the literature. The other two fusions have been described in other malignancies but not in melanoma: CANT1::ETV4 in prostate cancer and CCDC6::RET in thyroid cancer. The putative chimeric proteins are depicted with the N-terminal fusion partner shown in black, and the C-terminal partner in white. The exon breakpoints, listed on the right, are based on the following transcripts: CANT1 (ENST00000392446.5); ETV4 (ENST00000319349.5); CCDC6 (ENST00000263102.6); RET (ENST00000355710.3); NAGS (ENST00000293404.3); MAST2 (ENST00000361297.2). Relevant functional domains: DNA binding domain (ETS: erythroblast transformation specific), kinase domains (TKD: tyrosine kinase domain; MAST: microtubule-associated serine-threonine kinase; PKD: protein kinase domain; AGC: cAMP-dependent, cGMP-dependent and protein kinase C kinase C-terminal), WD: WD (tryptophan and aspartic acid) repeat domains, CC: coiled coil homodimerization domain; PDZ: PSD-95, Dlg1, Zo-1 protein interaction domain. The NOTCH1::GNB1 rearrangement, which has not been previously reported in the literature, was excluded from this figure because it is not expected to lead to the production of an in-frame chimeric fusion protein involving NOTCH1 and GNB1. As noted in the text, this rearrangement may result in the expression of the full length GNB1 protein under the control of the NOTCH1 promoter.
Additional fusions involving ALK (EML4::ALK, MLPH::ALK), BRAF (AGAP3::BRAF, AGK::BRAK, CDH3::BRAF, CCT8::BRAF, DIP2B::BRAF), RAF1 (EFNB1::RAF1, LRCH3::RAF1, MAP4::RAF1, RUFY1::RAF1), and TERT (ADCY2::TERT) have been previously reported in melanomas11,12,13,14,15,17,23,25,26.
Mutational analyses were successfully performed in samples from 15/16 patients with fusion-positive melanomas (Supplementary Table 2). For 2 patients, molecular testing was done on two available samples, and the same fusion and similar mutational profiles were detected in each specimen (Fig. 1). In one patient, the same AGK::BRAF fusion was detected in the primary tumor and in a bone metastasis resected one month later (Table 1, case 2). The two specimens were positive for the same mutations in NF1, KDR and in the TERT promoter, and the bone metastasis also showed homozygous loss of CDKN2A (Fig. 1, Supplementary Table 2). In another patient, the same RUFY1::RAF1 fusion was detected in two different metastases (lung and bone) that were resected 9 months apart (Table 1, case 9), and the two samples harbored the same mutations in PIK3CA, MEN1 and in the TERT promoter, and homozygous deletion of CDKN2A (Fig. 1, Supplementary Table 2). Fusion positive melanomas harbor oncogenic mutations in genes involved in receptor tyrosine kinase, MAPK/ERK, and mTOR signaling pathways (including ERBB4, MET, MAP2K1, PIK3CA, STK11, and TSC1). The most prevalent genetic abnormalities detected in our fusion-positive cohort involved the TERT gene (Fig. 1), and included 11/15 (73%) cases with hotspot mutations in the TERT promoter (C250T, 6 cases; C228T, 5 cases) and a TERT gene amplification in the sample harboring the ADCY2::TERT fusion (case 15). TERT promoter mutations were detected in 63% of fusion negative melanomas, and we found no significant association between the presence of a TERT promoter mutation and fusion positivity (p = 0.59) (Supplementary Table 3). Pathogenic mutations in CDKN2A, especially homozygous gene deletion, were the second most common finding, being detected in 3/15 (20%) patients with fusion-positive melanoma (Fig. 1). Gene fusions were largely mutually exclusive from mutations in the most common oncogenic drivers in melanoma. Pathogenic mutations in RAS (NRAS, KRAS or HRAS) family members, NF1 or KIT were absent from fusion-positive cases, while fusion-negative melanomas harbored mutations in all of them, at the following frequencies: 28% NRASmut, 31% RAS (NRAS, KRAS or HRAS)mut, and 6% KITmut. BRAF mutations were detected in 37% of fusion-negative melanomas and in 1/15 (7%) fusion positive cases.
There was a highly significant association between the presence of fusions and the absence of BRAF/RAS mutation (p < 0.00001, Supplementary Table 3), with gene fusions being detected in 5.4% of BRAF and RAS wild-type melanomas, and only in 0.2% of BRAF-mutant or RAS-mutant tumors. Our cohort included 166 (23%) triple wild-type melanomas. Interestingly, while TERT promoter mutations were very prevalent in fusion positive melanomas (11 of 15, 73%) and in BRAF/NRAS/NF1-driven cases (393 of 542, 73%), they were underrepresented in triple wild-type melanomas (56 of 166, 34%). Therefore, while gene fusions are very rare in melanoma (16 of 708, 2% in our cohort), they were significantly enriched within the subset of triple wild-type melanomas that harbor TERT promoter mutations (10 of 56, 18%, p < 0.00001, Supplementary Table 3).
Tumor mutational burden (TMB) was available for 412 cases. In our cohort, 48% (189 of 397) of fusion-negative tumors had a high TMB, while only 20% (3 of 15) of fusion-positive melanomas were found to have a high TMB (Table 2). High TMB cases included three cutaneous melanomas, positive for BRAF, RAF1 and NOTCH1 fusions (Table 1, cases 1, 6, and 14, respectively). Interestingly, for the 2 cases with very high TMB scores (cases 1 and 14, with TMB > 20), C > T transitions accounted for 73–78% of the mutations, a pattern consistent with exposure to UV radiation.
Fluorescence in situ hybridization, immunohistochemistry and immunofluorescence studies
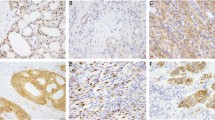
Selected rearrangements were confirmed by orthogonal assays. A commercially available NOTCH1 break apart FISH probe detected a NOTCH1 rearrangement in the melanoma sample harboring the novel NOTCH1::GNB1 fusion (case 14). The CDH3::BRAF and CCT8::BRAF fusions were confirmed by BRAF break apart FISH, and the EML4::ALK and MLPH::ALK rearrangements were validated by ALK break apart FISH (Fig. 3).

Fluorescence in-situ hybridization using break-apart probes for (A) ALK (case 9, MLPH::ALK) and (B) BRAF (case 4, CCT8::BRAF). Fusion-positive nuclei show splitting of the green and orange probe signals further than two probes apart (arrows), in addition to the normal un-split green-orange signal.
ALK immunostain performed on the melanoma harboring the EML4::ALK fusion (case 10) was positive for ALK expression (Fig. 4). To evaluate the effects of previously unreported rearrangements, we performed immunoassays on fusion-positive melanomas and on a fusion-negative melanoma harboring a BRAF V600E mutation, which is the most common genetic alteration observed in these tumors (Fig. 5). We also assessed publically available datasets, to examine protein expression levels across multiple melanomas (Supplementary Figs. 1–3). Notch1 immunostaining demonstrated that the melanoma sample harboring the NOTCH1::GNB1 rearrangement (case 14) was positive for Notch1 expression (Fig. 5A), and that Notch1 protein levels were comparable to the ones observed in a BRAF V600E-mutant melanoma (Fig. 5B). Data from The Human Protein Atlas demonstrated weak (6 of 12) and moderate (6 of 12) Notch1 expression across 12 melanoma samples, with most cases (11 of 12) exhibiting a cytoplasmic/membranous and nuclear distribution of the protein (Supplementary Fig. 1).

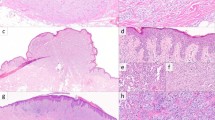
An epithelioid morphology is seen in melanomas with A, D CANT1 (case 12), B, E BRAF (case 3), C, F MAST2 (case 16), and G NOTCH1 (case 14) fusions; whereas a spindle morphology is seen in a melanoma with H RAF1 fusion (case 8). I ALK immunostain is strongly positive in the melanoma with ALK fusion (case 10).

Notch1 immunostains demonstrated similar levels of Notch1 protein expression in (A) a melanoma harboring a NOTCH1::GNB1 rearrangement and in B a BRAF V600E-mutant melanoma. Immunofluorescence using a GNB1-specific antibody (C, D) detected uniform cytoplasmic and membranous GNB1 expression in (C) a melanoma harboring a NOTCH1::GNB1 rearrangement (case 14), and heterogeneous distribution of GNB1 in (D) a BRAF V600E-mutant melanoma. Immunofluorescence using a MAST2-specific antibody (E, F) showed similar patterns of heterogeneous distribution of MAST2 protein in (E) a melanoma harboring a NAGS::MAST2 rearrangement (case 16) and in (F) a BRAF V600E-mutant melanoma.
Immunofluorescence staining using a GNB1-specific antibody demonstrated uniform cytoplasmic and membranous GNB1 expression in the NOTCH1::GNB1 positive melanoma (case 14) (Fig. 5C), and heterogeneous distribution of GNB1 in a BRAF V600E-mutant melanoma (Fig. 5D). GNB1 immunostaining, made available by The Human Protein Atlas, showed variable levels of cytoplasmic/membranous GNB1 expression across 11 melanomas, ranging from moderate (4 of 11) and weak (3 of 11), to no detectable levels in 4 of 11 cases (Supplementary Fig. 2). Immunofluorescence analysis using a MAST2-specific antibody showed similar patterns of heterogeneous expression of MAST2 in the melanoma sample harboring a NAGS::MAST2 rearrangement (case 16) (Fig. 5E) and in a BRAF V600E-mutant melanoma (Fig. 5F). MAST2 immunostaining results from The Human Protein Atlas, demonstrated variable levels of cytoplasmic/membranous MAST2 in 12 melanomas, with most cases showing moderate (3 of 12) or weak (5 of 12) expression, and 4 of 12 cases being negative for MAST2 (Supplementary Fig. 3).
Clinical and histological features
The primary tumor site of all analyzed melanomas included 599 (80%) cutaneous, 38 (5%) acral, 11 (1.5%) anorectal, 23 (3%) sinonasal, 27 (3.6%) eye (uveal/conjunctiva), 11 (1.5%) genital (vulva/penile), and 41 (5.5%) cases of unknown primary. Within the fusion-positive cohort, 12/599 (2%) melanomas were cutaneous, 2/38 (5%) acral, 1/9 (11%) vulvar, and 1/23 (4.3%) sinonasal (Table 1). MLPH::ALK and ADCY2::TERT fusions involved the acral site, CANT1::ETV4 the vulva, EML4::ALK the sinonasal region, and the remaining fusions were detected in in tumors from cutaneous sites. We found no significant difference between the prevalence of fusions in cutaneous (2.2%) versus non-cutaneous (3.6%) sites (Supplementary Table 3).
The sites of the 12 metastases were lung (3), lymph node (3), bone (2), brain (2), skin (1) and soft tissue (1). Overall, there were 438 males and 312 females, including 427/734 (58%) males with fusion-negative tumors and 11/16 (69%) male patients with fusion positive melanomas. There was no significant association between gender and fusion positivity (Supplementary Table 3). The age at diagnosis ranged from 29 to 83 years (median, 56 years). Two (12.5%) patients presented with stage IV disease (distant metastases), 8 (50%) with stage III (nodal involvement), 5 (31%) with stage II, and 1 (6%) patients with stage I melanoma (Table 1)31.
The majority of fusion positive cases (92%, 12 of 13) exhibited an epithelioid morphology, with only one tumor (positive for a MAP4::RAF1 fusion) demonstrating spindle cell morphology (Fig. 4). Surgical specimens were available for histopathological review in 4 out of 5 BRAF fusion-positive melanomas, and none of them exhibited a spindled morphology.
Treatment and outcome
Fifteen of 16 patients (94%) underwent surgery, primarily for the treatment of localized disease (to excise the primary tumor and nodal metastases). Two patients received surgery alone and have no evidence of disease after 13 and 25 months. Seven patients were treated with radiotherapy. Two patients enrolled in clinical trials using cryoablation to target metastases.
Thirteen (81%) patients were treated with immunotherapy for metastatic disease or in the adjuvant setting. Nine and eight patients received the PD-1 inhibitors pembrolizumab and nivolumab, respectively and nine patients received anti-CTLA-4 therapy (ipilimumab). Nivolumab was often used in combination with other agents (on 6 occasions with ipilimumab, and once with: CCR2 antagonist TAK-202, anti-LAG3 antibody, and T-VEC oncolytic virus). Nine patients received immunosuppressive treatment, in most cases, to manage serious adverse events associated with immune checkpoint inhibitor therapy.
Six of 12 (50%) patients with potentially actionable fusions progressed on immunotherapy, and 3/6 (50%) were treated with targeted agents (ALK and MET inhibitors), 2 off-label and 1 as part of a clinical trial. One patient with an AGAP3::BRAF fusion positive melanoma experienced a 30-month long response to trametinib. Another patient whose tumor harbored a RUFY1::RAF1 fusion was placed on binimetinib, a MEK inhibitor. Treatment only lasted four months, as the patient experienced a mixed response to this MEK inhibitor, with major response to therapy in his visceral organs but marked progression in his bone metastases. One patient whose melanoma harbored an EML4::ALK fusion received an ALK inhibitor (alectinib), but experienced disease progression after one month (Table 1). Follow-up since time of diagnosis ranges from 12 months to 173 months (median, 41 months). Six patients died and ten are alive (4 with disease and 6 with no evidence of disease) (Table 1).
Discussion
We identified 4 novel fusions, including 2 that have been reported in other malignancies but not in melanoma (CANT1::ETV4 and CCDC6::RET) and 2 fusions that have not been described in the literature (NAGS::MAST2 and NOTCH1::GNB1). A CANT1::ETV4 fusion was detected in one primary vulvar melanoma in our series. To date, CANT1::ETV4 fusion has only been reported in prostate carcinoma32. ETV4 belongs to the family of ETS-related transcription factors and has been reported as a rare fusion partner of EWSR1 in classical Ewing sarcoma33. Calcium-activated nucleotidase 1 (CANT1) expression is regulated by androgen and its exon 1a transcript is preferentially expressed in the prostate32. A CCDC6::RET fusion was detected in a primary melanoma from the lower extremity in our series. The same fusion between CCDC6 exon 1 and RET exon 12 has been previously documented in papillary thyroid carcinoma, but not in melanoma34. NAGS::MAST2 and NOTCH1::GNB1 rearrangements, identified in two metastatic melanomas in our series, have not been reported in any malignancies. Recurrent rearrangements of the microtubule associated serine-threonine (MAST) kinase and NOTCH gene families have been reported in breast cancer35. Breast cancer cell lines harboring NOTCH rearrangements have been shown to exhibit sensitivity to inhibition of Notch signaling35. In addition, overexpression of MAST1 or MAST2 fusions had a proliferative effect on tumor cells35.
Our sequencing results do not support the production of an in-frame NOTCH1::GNB1 chimeric protein, suggesting that (in contrast to the fusions reported in breast cancer) this rearrangement may result in loss of function of NOTCH1. Interestingly, immunohistochemistry showed that the presence of this rearrangement does not seem to reduce Notch1 protein levels in the NOTCH1::GNB1-positive tumor, when compared to a BRAF V600E-positive melanoma (Fig. 5A, B). The results suggest that, although the rearrangement may inactivate one copy of NOTCH1, it does not appear to significantly impact NOTCH1 activity in this tumor (presumably because the second allele remains intact). As noted, the NOTCH1 rearrangement juxtaposes NOTCH1 exon 2 immediately upstream of the GNB1 starting codon and it could, potentially, drive GNB1 expression under the control of the NOTCH1 promoter. GNB1 encodes the beta subunit of a trimeric G protein complex that mediates signaling downstream of G-protein coupled receptors. Previous research showed that GNB1 is activated by recurrent somatic mutations in hematopoietic malignancies and in solid tumors, including melanoma36. A recent study showed that GNB1 is overexpressed in a subset of cervical squamous cell carcinomas, with high levels of GNB1 being associated with a poor prognosis37. Data from The Human Protein Atlas indicate that approximately 40% of melanomas are negative for GNB1, while ~60% of cases exhibit weak to moderate expression of the protein (Supplementary Fig. 2). Given the strong, uniform distribution of GNB1 observed in the NOTCH1::GNB1-positive melanoma (Fig. 5C), we hypothesize that the rearrangement may be upregulating GNB1 expression in this tumor, under the control of the NOTCH1 promoter.
MAST2 encodes a microtubule-associated serine/threonine kinase with anti-apoptotic properties, that is overexpressed in various tumors, including esophageal cancer, pancreatic cancer, and sarcomas38. In liver cancer, high expression of MAST2 has been associated with advanced clinical stage and with a poor prognosis39. In addition, overexpression of a MAST2 fusion detected in breast cancer had a proliferative effect on tumor cells35. The fusion described in that study had a different N-terminal partner from our melanoma case, but it involved the same MAST2 breakpoint (exon 5) as the NAGS::MAST2 rearrangement. We detected a similar pattern of MAST2 expression in the NAGS::MAST2 -positive sample and in a BRAF V600E-mutant melanoma (Fig. 5E, F). Publically available datasets indicate that MAST2 is expressed in a subset of melanomas (Supplementary Fig. 3), suggesting that different tumors may induce MAST2 expression through different mechanisms. Taken together, our data suggest that the NAGS::MAST2 fusion observed in case 16 may function as another mechanism to drive MAST2 expression.
BRAF fusions are common kinase translocations in tumors40. Although estimated to occur in 2.6%–6.7% of all melanomas, the frequency of BRAF fusions is higher in melanomas occurring in younger females and in Spitz neoplasms23,41. Four of our five BRAF fusion-positive tumors with available histologic sections exhibit an epithelioid morphology, not a spindled morphology, similar to previous report25. Treatment response to MEK inhibitor in a patient with SKAP2::BRAF fusion has been described in a recent case report22. Similarly, one of our patients with an AGAP3::BRAF fusion positive melanoma experienced a 30-month long response to trametinib. This is in contrast with a previously reported AGAP3::BRAF fusion that conferred resistance in cell lines to a BRAF inhibitor in a patient with melanoma42. High expression of a BRAF fusion kinase has been shown to promote resistance to chemotherapy41.
The presence of a RAF1 fusion in melanocytic neoplasm is rare, occurring in less than 1% of neoplasms40. BRAF and CRAF, encoded by RAF1, form a heterodimer which activate the MEK-ERK pathway resulting in cell proliferation and survival43. Cell lines with RAF1 fusions exhibit increased MEK phosphorylation in comparison to wild type44. RAF and MEK inhibitors may be useful in a subset of gene fusion-harboring solid tumors12,19,21. Excellent clinical responses were documented in 2 patients with RAF1-fusion melanomas following immunotherapy failure19,21. Four RAF1 (EFNB1::RAF1, LRCH3::RAF1, MAP4::RAF1, RUFY1::RAF1) fusions were detected in four melanoma metastases in our series. In one patient, the same RUFY1::RAF1 fusion was detected in two metastatic tumors. The patient was treated with a MEK inhibitor for four months. He experienced major response to therapy in his visceral organs but had marked progression in his bone metastases, possibly due to tumor heterogeneity or to impaired drug access to the bone lesions. Previously reported RAF1 fusion partners include AGGF1, ANO10, CDH3, CLCN6, CTDSPL, CTNNA1, GOLGA4, LMNA, LRCH3, LRRFIP2, MAP4, MPRIP, PAPD7, PRKAR2A, SASS6, SOX5, and TRAK112,14,19,21,40,45,46,47. Consistent with prior studies, the four melanomas with RAF1 fusion were negative for pathogenic mutations in BRAF, NRAS and NF1 (Fig. 1, Supplementary Table 2)14,47. TERT and CDKN2A concomitant mutations are frequently noted in the literature14. Similar to our findings, the majority of melanomas with RAF1 fusion exhibit an epithelioid morphology14.
ALK fusions are detected in 8–11%, 5–16% and 1–3% of Spitz nevi, atypical Spitz tumors and 1–3% Spitz melanomas, respectively17,48. Melanophilin (MLPH) gene was reported as partner gene to ALK in two Spitz nevi49,50. Echinoderm microtubule-associated protein-like 4 (EML4) is another partner gene to ALK20,51. Although the patient with the EML4::ALK fusion (case 10) did not respond to treatment with alectinib, a separate case report documented successful ALK inhibitor treatment for carcinoma with EML4::ALK fusion, suggesting the potential for ALK inhibitor use in melanoma52.
TERT aberrations are detected most frequently in acral melanoma as compared with other melanoma subtypes53. In the fusion-positive melanomas in our series, TERT promoter mutation is the most frequent mutation identified, especially TERT 250 and TERT 228, followed by, recurrent inactivation of CDKN2A. Mutational analyses identified 14/15 triple wild-type tumors (negative for pathogenic mutations in BRAF, RAS and NF1) tumors.
Notably, nearly all tumors with gene fusions were BRAF and RAS wild-type (14/15; 93%) with one tumor with a CCDC6::RET fusion harboring two BRAF mutations in cis: the highly prevalent BRAF V600E mutation and a BRAF W604C variant of uncertain significance. It is unclear if, in this tumor, the proximity of the BRAF W604C variant affects the pathogenicity of the BRAF V600E mutation. Although concurrent RET fusions and BRAF mutations have been reported in papillary thyroid carcinoma (reported in 2.5–19.4% of tumors), gene fusions and BRAF and NRAS mutations are thought to be mutually exclusive in melanoma in the vast majority of cases54. Overall, our findings are consistent with previous literature that report that melanomas with fusions often lack common driver mutations (BRAF, RAS and NF1)13. Gao et al. have investigated fusions in 9624 tumors of 33 different cancer types of The Cancer Genome Atlas (TCGA) cohort and reported that mutations were not found when fusion was already present in the same gene, supporting their mutual exclusivity in many malignancies55. We noted that, in our series, gene rearrangements were significantly enriched within the subset of triple wild-type melanomas that harbor TERT promoter mutations (18%). Since many cancer genotyping panels cover BRAF, NRAS, NF1 and TERT promoter mutations, if a melanoma sample is “triple wild-type” and positive for a TERT promoter mutation, it may be worth testing it for gene rearrangements. In our cohort, 9 of those 10 (triple wild-type, TERT-mutant, fusion-positive) melanomas harbored a potentially actionable fusion. Identification of a fusion in such cases may be of critical importance as targeted therapy may provide an additional treatment option for clinicians. In our study, one patient with a BRAF fusion-positive melanoma experienced a durable response to treatment with a MEK inhibitor (case 1), while two other patients showed mixed responses to selective inhibitors (cases 9 and 10). Although more studies are needed to better understand the factors that regulate treatment effectiveness, the use of targeted therapy has been reported to improve patient survival in some case reports22.
In summary, we reported four novel fusions in melanomas: 2 fusions have been reported in other malignancies but not in melanoma and 2 have not been described in the literature. Our study shows a very strong correlation between the presence of a fusion and the lack of BRAF/RAS/NF1 driver mutations, especially in TERT promoter-mutant melanomas. Detecting fusions in patients with advanced melanoma may have a clinically significant impact, especially in patients triple wild-type melanomas that harbor TERT promoter mutations, who have failed front-line immunotherapy.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Matthews, N.H., Li, W.Q., Qureshi, A.A., Weinstock, M.A., Cho, E. Epidemiology of Melanoma. In: Ward WH, Farma JM, editors. Cutaneous Melanoma: Etiology and Therapy [Internet]. Brisbane (AU): Codon Publications (2017). Chapter 1. Available from: https://www.ncbi.nlm.nih.gov/books/NBK481862/https://doi.org/10.15586/codon.cutaneousmelanoma.2017.ch1.
U.S. Cancer Statistics Working Group. United States Cancer Statistics: 1999–2010 Incidence and Mortality Web-based report. Atlanta, GA: Centers for Disease Control and Prevention, U.S. Dept of Health and Human Services and National Cancer Institute, National Institutes of Health (2013).
Chapman, P.B., Hauschild, A., Robert, C., Haanen, J.B., Ascierto, P., Larkin, J. et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 364, 2507–16 (2011).
Yang, K., Oak, A.S.W., Slominski, R.M., Brożyna, A.A., Slominski, A.T. Current molecular markers of melanoma and treatment targets. Int. J. Mol. Sci. 21, 3535 (2020). https://doi.org/10.3390/ijms21103535.
Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell. 161, 1681–1696 (2015).
Huang, F.W., Hodis, E., Xu, M.J., Kryukov, G.V., Chin, L., Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science. 339, 957–9 (2013).
Yan, X., Larsson, C., Xu, D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: old actors and new players. Oncogene. 38, 6172–6183 (2019).
Craig, S., Earnshaw, C.H., Virós, A. Ultraviolet light and melanoma. J. Pathol. 244, 578–585 (2018).
Vergara, I.A., Wilmott, J.S., Long, G.V., Scoyler, R.A. Genetic drivers of non-cutaneous melanomas: challenges and opportunities in a heterogeneous landscape. Exp. Dermatol. 31, 13–30 (2021).
Mertens, F., Johansson, B., Fioretos, T., Mitelman, F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer. 15, 371–81 (2015).
Yoshihara, K., Wang, Q., Torres-Garcia, W., Zheng, S., Vegesna, R., Kim, H., Verhaak, R.G. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene. 34, 4845–54 (2015).
Palanisamy, N., Ateeq, B., Kalyana-Sundaram, S., Pflueger, D., Ramnarayanan, K., Shankar, S. et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat. Med. 16, 793–798 (2010).
Turner, J., Couts, K., Sheren, J., Saichaemchan, S., Ariyawutyakorn, W., Avolio, I. et al. Kinase gene fusions in defined subsets of melanoma. Pigment. Cell. Melanoma. Res. 30, 53–62 (2017).
Williams, E.A., Shah, N., Montesion, M., Sharaf, R., Pavlick, D.C., Sokol, E.S. et al. Melanomas with activating RAF1 fusions: clinical, histopathologic, and molecular profiles. Mod. Pathol. 33, 1466–1474 (2020).
Forschner, A., Forschhammer, S., Bonzheim, I. NTRK gene fusions in melanoma: detection, prevalence and potential therapeutic implications. J. Dtsch. Dermatol. Ges. 18, 1387–1392 (2020).
Quan, V.L., Panah, E., Zhang, B., Shi, K., Mohan, L.S., Gerami, P. The role of gene fusions in melanocytic neoplasms. J. Cutan. Pathol. 46, 878–887 (2019).
Shaw, A.T., Gandhi, L., Gadgeel, S., Riely, G.J., Cetnar, J., West, H. et al. Alectinib in ALK-positive, crizotinib-resistant, non-small-cell lung cancer: a single-group, multicentre, phase 2 trial. Lancet. Oncol. 17, 234–242 (2016).
Menzies, A.M., Yeh, I., Botton, T., Bastian, B.C., Scoyler, R.A., Long, G.V. Clinical activity of the MEK inhibitor trametinib in metastatic melanoma containing BRAF kinase fusion. Pigment. Cell. Melanoma. Res. 28, 607–610 (2015).
Kim, K.B., Semrad, T., Schrock, A.B., Ali, S.M., Ross, J.S., Singer, M. et al. Significant clinical response to a MEK inhibitor therapy in a patient with metastatic melanoma harboring an RAF1 fusion. JCO. Precis. Oncol. 2, 1–3 (2018).
Couts, K.L., Bemis, J., Turner, J.A., Bagby, S.M., Murphy, D., Christiansen, J. et al. ALK inhibitor response in melanomas expression EML4-ALK fusions and alternate ALK isoforms. Mol. Cancer. Ther. 17, 222–231 (2018).
McEvoy, C.R., Xu, H., Smith, K., Etemadmoghadam, D., Leong, H.S., Choong, D.Y. et al. Profound MEK inhibitor response in a cutaneous melanoma harboring a GOLGA4-RAF1 fusion. J. Clin. Invest. 129, 1940–1945 (2019).
Chew, S.M., Lucas, M., Brady, M., Kelly, C.M. SKAP2-BRAF fusion and response to an MEK inhibitor in a patient with metastatic melanoma resistant to immunotherapy. BMJ. Case. Rep. 14, e238494 (2021). https://doi.org/10.1136/bcr-2020-238494.
Wiesner, T., He, J., Yelensky, R., Esteve-Puig, R., Botton, T., Yeh I. et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat. Commun. 5, 3116 (2014). https://doi.org/10.1038/ncomms4116.
Ferrara, G., Gianotti, R., Cavicchini, S., Salviato, T., Zalaudek, I., Argenziano, G. Spitz nevus, Spitz tumor, and Spitzoid melanoma: a comprehensive clinicopathologic overview. Dermatol. Clin. 31, 589–598 (2013).
Kim, D., Khan, A.U., Compres, E.V., Zhang, B., Sunshine, J.C., Quan, V.L. et al. BRAF fusion Spitz neoplasms; clinical morphological, and genomic findings in six cases. J. Cutan. Pathol. 47, 1132–1142 (2020).
Niu, H.T., Zhou, Q.M., Wang, F., Shao, Q., Guan, Y.X., Wen, X.Z. et al. Identification of anaplastic lymphoma kinase break points and oncogenic mutation profiles in acral/mucosal melanomas. Pigment. Cell. Melanoma. Res. 26, 646–53 (2013).
Zheng, Z., Liebers, M., Zhelyazkova, B., Cao, Y., Panditi, D., Lynch, K.D. et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat. Med. 20, 1479–1484 (2014).
Li, H., Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25, 1754–60 (2009).
Kent, W.J. BLAT – the BLAST-like alignment tool. Genome Res 12, 656–64 (2002).
Thierauf, J., Ramamurthy, N., Jo, V.Y., Robinson, H., Frazier, R.P, Gonzalez, J. et al. Clinically integrated molecular diagnostics in adenoid cystic carcinoma. Oncologist. 24, 1356–67 (2019).
Keung, E.Z., Gershenwald, J.E. The eighth edition American Joint Committee on Cancer (AJCC) melanoma staging system: implications for melanoma treatment and care. Expert. Rev. Anticancer. Ther. 18, 775–784 (2018).
Hermans, K.G., Bressers, A.A., van der Korput, H.A., Dits, N.F., Jenster, G., Trapman, J. Two unique novel prostate-specific and androgen-regulated fusion partners of ETV4 in prostate cancer. Cancer. Res. 68, 3094–8 (2008).
Kanedo, Y., Yoshida, K., Handa, M., Toyoda, Y., Nishihira, H., Tanaka, Y. et al. Fusion of an ETS-family gene, EIAF, to EWS by t(17;22)(q12;q12) chromosome translocation in an undifferentiated sarcoma of infancy. Genes. Chromosomes. Cancer. 15, 115–121 (1996).
Celestino, R., Sigstad, E., Lovf, M., Thomassen, G.O.S., Groholt, K.K., Jorgensen, L.H. et al. Survey of 548 oncogenic fusion transcripts in thyroid tumours supports the importance of the already established thyroid fusions genes. Genes. Chromosomes. Cancer. 51, 1154–1164 (2012).
Robinson, D.R., Kalyana-Sundaram, S., Wu, Y.M, Shankar, S., Cao, X., Ateeq, B. et al. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat. Med. 17, 1646–1651 (2012).
Yoda, A., Adelmant, G., Tamburini, J., Chapuy, B., Shindoh, N., Yoda, Y. et al. Mutations in G protein beta subunits promote transformation and kinase inhibitor resistance. Nature. 21, 71–5 (2015).
Cao, Y., Li, J., Jia, Y., Zhang, R., Shi, H. Circ RNA circ_POLA2 promotes cervical squamous cell carcinoma progression via regulating miR-326/GNB1. Front. Oncol. 10, 959 (2020) https://doi.org/10.3389/fonc.2020.00959.
Eißmann, M., Schwamb, B., Melzer, I.M., Moser, J., Siele, D., Kohl, U. et al. A functional yeast survival screen of tumor-derived cDNA libraries designed to identify anti-apoptotic mammalian oncogenes. PLoS. One. 8, e64873 (2013) https://doi.org/10.1371/journal.pone.0064873.
Jiao, Y., Li, Y., Jiang, P., Fu, Z., Liu, Y. High MAST2 mRNA expression and its role in diagnosis and prognosis of liver cancer. Sci. Rep. 9, 19865 (2019) https://doi.org/10.1038/s41598-019-56476-x.
Stransky, N., Cerami, E., Schalm, S., Kim, J.L., Lengauer, C. The landscape of kinase fusions in cancer. Nat. Commun. 5, 4846 (2014). https://doi.org/10.1038/ncomms5846.
Botton, T., Talevich, E., Mishra, V.K., Zhang, T., Shain, A.H., Berquet, C. et al. Genetic heterogeneity of BRAF fusion kinases in melanoma affects drug responses. Cell. Rep. 29, 573–588 (2019).
Kulkarni, A., Al-Hraishawi, H., Simhadri, S., Hirshfield, K.M., Chen, S., Pine, S. et al. BRAF fusion as a novel mechanism of acquired resistance to Vemurafenib in BRAFV600E mutation melanoma. Clin. Cancer. Res. 23, 5631–5638 (2017).
Lavole, H., Therrien, M. Regulation of RAF protein kinases in ERK signaling. Nat. Rev. Mol. Cell. Biol. 16, 281–298 (2015).
Jones, D.T., Kocialkowski, S., Liu, L., Pearson, D.M., Ichimura, K., Collins, V.P. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene. 28, 2119–2123 (2009).
Hayward, N.K., Wilmott, J.S., Waddell, N., Johansson, P.A., Field, M.A., Nones, K., et al. Whole-genome landscapes of major melanoma subtypes. Nature. 545, 175–180 (2017).
Baltres, A., Salhi, A., Houlier, A., Pissaloux, D., Tirode, F., Haddad, V. et al. Malignant melanoma with areas of rhabdomyosarcomatous differentiation arising in a giant congenital nevus with RAF1 gene fusion. Pigment. Cell. Melanoma. Res. 32, 708–713 (2019).
LeBlanc, R.E., Lefferts, J.A., Baker, M.L., Linos, K.D. Novel LRRFIP2-RAF1 fusion identified in an acral melanoma: a review of the literature on melanocytic proliferations with RAF1 fusions and the potential therapeutic implications. J. Cutan. Pathol. 47, 1181–1186 (2020).
Wiesner, T., Kutzner, H., Cerroni, L., Mihm Jr, M.C., Busam, K.J., Murali, R. Genomic aberrations in Spitzoid melanocytic tumors and their implications for diagnosis, prognosis and therapy. Pathology. 48, 113–31 (2016).
Chung, C.T., Marrano, P., Swanson, D., Dickson, B.C., Thorner, P.S. Fusion of ALK to the melanophilin gene MLPH in pediatric Spitz nevi. Hum. Pathol. 87, 57–64 (2019).
Fujimoto, M., Togashi, Y., Matsuzaki, I., Baba, S., Takeuchi, K., Inaba, Y. et al. A case report of atypical Spitz tumor harboring a novel MLPH-ALK gene fusion with discordant ALK immunohistochemistry results. Hum. Pathol. 80, 99–103 (2018).
Salari, B., Duncan, L.M., Lennerz, J.K., Holbrook, E.H., Emerick, K.S., Foreman, R.K. Observed progression from melanosis with melanocyte hyperplasia to sinonasal melanoma with distant metastasis and a unique genetic rearrangement. J. Cutan. Pathol. 48, 948–953 (2021).
Sugiyama, K., Izumika, A., Iwakoshi, A., Nishibori, R., Sato, M., Shiraishi, K., et al. Successful Alectinib treatment for carcinoma of unknown primary with EML4-ALK fusion gene: a case report. Curr. Oncol. 28, 1938–1945 (2021).
Liang, W.S., Hendricks, W., Kiefer, J., Schmidt, J., Sekar, S., Carpten, J. et al. Integrated genomic analyses reveal frequent TERT aberrations in acral melanoma. Genome. Res. 27, 524–532 (2017).
Zhang, R., Dong, L., Yu, J. Concomitant pathogenic mutations and fusions of driver oncogenes in tumors. Front. Oncol. 10, 544579 (2021). https://doi.org/10.3389/fonc.2020.544579.
Gao, Q., Liang, W.W., Foltz, S.M., Mutharasu, G., Jayasinghe, R.G., Cao, S., et al. Driver fusions and their implications in the development and treatment of human cancers. Cell. Rep. 23, 227–238.e3 (2018).
Author information
Authors and Affiliations
Contributions
Conceptualization and supervision: D.D.S. and M.P.H; methodology: L.P.L., V.N., J.G., A.A.F., S.S., M.L.O., J.K.L., D.D.S. and M.P.H; acquisition, analysis and interpretation of data: J.M.T.M., L.P.L., V.N., J.G., A.A.F., S.S., M.L.O., R.K.F., L.M.D., D.P.L., J.K.L., D.D.S. and M.P.H; writing original draft: J.M.T.M., D.D.S. and M.P.H; review and edit manuscript: J.M.T.M., L.P.L., V.N., J.G., A.A.F., S.S., M.L.O., R.K.F., L.M.D., D.P.L., J.K.L., D.D.S and M.P.H. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Partners Human Research Committee, the institutional review board of Partners HealthCare (protocol code 2011P0001665).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Moran, J.M.T., Le, L.P., Nardi, V. et al. Identification of fusions with potential clinical significance in melanoma. Mod Pathol 35, 1837–1847 (2022). https://doi.org/10.1038/s41379-022-01138-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-022-01138-z
This article is cited by
-
Therapeutic Strategies in BRAF V600 Wild-Type Cutaneous Melanoma
American Journal of Clinical Dermatology (2024)
-
Multiple drugs
Reactions Weekly (2023)
-
Translocations and Gene Fusions in Sinonasal Malignancies
Current Oncology Reports (2023)
