
Abstract
Regulatory T cells (Tregs) mediate immune homeostasis, yet also facilitate nuanced immune responses during infection, balancing pathogen control while limiting host inflammation. Recent studies have identified Treg populations in non-lymphoid tissues that are phenotypically distinct from Tregs in lymphoid tissues (LT), including performance of location-dependent roles. Mucosal tissues serve as critical barriers to microbes while performing unique physiologic functions, so we sought to identify distinct phenotypical and functional aspects of mucosal Tregs in the female reproductive tract. In healthy human and mouse vaginal mucosa, we found that Tregs are highly activated compared to blood or LT Tregs. To determine if this phenotype reflects acute activation or a general signature of vaginal tract (VT)-residency, we infected mice with HSV-2 to discover that VT Tregs express granzyme-B (GzmB) and acquire a VT Treg signature distinct from baseline. To determine the mechanisms that drive GzmB expression, we performed ex vivo assays to reveal that a combination of type-I interferons and interleukin-2 is sufficient for GzmB expression. Together, we highlight that VT Tregs are activated at steady state and become further activated in response to infection; thus, they may exert robust control of local immune responses, which could have implications for mucosal vaccine design.

Similar content being viewed by others


Introduction
CD4+ regulatory T cells (Tregs), defined by the expression of transcription factor forkhead box P3 (FoxP3), are crucial for maintaining tolerance and preventing aberrant inflammation1,2,3,4,5. Early Treg studies focused on their development in the thymus and Tregs in lymphoid organs, but more recently, Treg subsets have been identified in discrete tissue compartments such as hair follicles, skin, muscle, and visceral adipose tissue (VAT)6,7,8,9,10,11,12,13,14. Although these tissue Treg populations share hallmarks of tissue residency, these populations have distinct transcriptional profiles, phenotypes, and clonally expanded T cell receptor (TCR) repertoires that do not overlap with lymphoid Treg counterparts or conventional T cells from the same tissue compartment7,8,14,15,16. Additionally, tissue Tregs carry out location-specific functions outside of their canonical suppressive roles, such as directly contributing to muscle tissue healing by producing growth factor amphiregulin (Areg)6,7 or facilitating adipocyte metabolism in the VAT8,9,10. Thus, Treg functions extend beyond classical suppressor roles and are highly location-dependent, suggesting that other tissue sites may host their own specialized Treg populations.
More recently, distinct Treg phenotypes and functions have also been identified in mucosal tissues. The mucosa serves as a barrier against pathogens as well as the site of specialized functions such as nutrient and air exchange, necessitating that Tregs orchestrate carefully controlled, specialized immune responses. In the small intestine and colon, both thymically derived and peripherally induced Tregs contribute to gut homeostasis by facilitating oral tolerance to dietary antigens, commensal bacteria, and preventing Inflammatory Bowel Diseases (IBD)17,18. Likewise, lung Treg populations are implicated in preventing allergy and airway inflammation19,20,21,22,23,24,25,26, and like muscle Tregs, have been shown to produce Areg in response to lung damage27. These discoveries suggest that specialized Tregs may be overseeing immune responses and performing yet-unknown functions in other mucosal tissues. Additionally, given their localization within mucosal sites, Tregs interact with local T cells15,28,29,30,31,32, including non-circulating tissue-resident memory T cells (Trm)33,34,35. Both CD4+ and CD8+ mucosal Trm are important controllers of local viral infections, including vaginal HSV-2 infection33,36,37,38,39,40. Therefore, muscoal STI vaccine design efforts are focused on eliciting robust Trm responses at the site of infection41,42. Given that Tregs are found alongside Trm in HSV-2 lesions43, vaginal Trm are likely subject to Treg-mediated control, but this relationship has not been previously studied within the vagina or during HSV-2 infection.
While Tregs have been extensively studied in the context of autoimmunity, the role of Tregs during infection is less defined and varies by infection32,44,45,46. In several infection models, Tregs limit or resolve immune-mediated tissue damage and tune effector T cell responses17,27,29,47,48,49, often at the expense of delayed pathogen clearance15,28,30,31. Notably, during vaginal infection with herpes simplex virus 2 (HSV-2), peripheral Tregs facilitate proper migration of antigen-bearing dendritic cells (DCs) to the vaginal-draining lymph node (dLN), and systemic Treg ablation causes a delay in antigen-specific T cell priming, associated with worsened viral-mediated disease50,51. Together, these findings demonstrate that in infectious contexts, Tregs serve as a fulcrum between immunity and tolerance, tasked with allowing for productive effector immune responses while also limiting collateral tissue damage caused by aggressive inflammatory responses.
Tregs have now been described in various mucosal tissues and infections52, but Tregs in the female genitourinary (GU) tract, including the uterus, cervix, and vaginal mucosa, are understudied in both health and infection. GU tissues are extraordinary in that they are the home of the tolerogenic processes of conception and pregnancy, but also the entry site of sexually transmitted infections (STIs). Therefore, the GU tract must facilitate tolerance to commensal bacteria, reproductive antigens, and the fetus, while also mounting early immune responses against invading infection. Although there is some evidence for uterine Treg-mediated tolerance during pregnancy53,54,55,56, GU tract Tregs, and more specifically vaginal tract (VT) Tregs, remain poorly characterized. Considering the paradoxical immunological requirements of the vagina, it is likely that Tregs are crucial in maintaining the tolerance:immunity axis in this mucosal site. Thus, we hypothesized that VT Tregs are a unique, tissue-resident population that perform specialized functions in homeostasis and infection. To test this, we used flow cytometry and RNA-sequencing (RNA-seq) to interrogate the previously uncharacterized vaginal Treg compartment at homeostasis in mouse and human tissues. We then compared baseline Treg phenotypes to Tregs during active, local viral infection in mice. Finally, we investigated the inflammatory signals that drive differential phenotypes in vaginal Tregs. Here, we demonstrate that vaginal mucosal Tregs in healthy mice and humans are more activated compared to circulating Tregs, suggesting that vaginal tissue localization alone confers a distinct phenotype on local Tregs. However, we also show that upon vaginal infection with HSV-2, vaginal Tregs become further activated, including the acquisition of robust, cytotoxic GzmB expression. We found that IL-2 in combination with type-I interferons (IFN-I) induce expression of GzmB on Tregs, uncovering the inflammatory cues that orchestrate Treg expression of GzmB. Overall, our findings demonstrate that the previously unstudied vaginal Treg population is constitutively activated in homeostasis but becomes further activated after infection, gaining expression of GzmB. These Tregs may be poised with cytoxic potential to regulate local inflammation, an important consideration for mucosal vaccine design.
Results
Vaginal mucosal Tregs in healthy women are activated compared to circulating Tregs
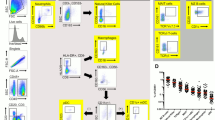
To phenotype human VT Tregs, we received vaginal mucosal biopsies and peripheral blood mononuclear cells (PBMC) from 14 healthy Seattle-area women without any known genital tract infections (Fig. 1a). Using flow cytometry, Tregs were identified as CD3+ CD4+ CD25+ CD127-FoxP3+, conventional CD4 T cells (CD4Tconv) as CD3+ CD4+ CD25−, and CD8 T cells as CD3+ CD8+ (Fig. 1b; Supplementary Fig. 1 and Supplementary Table 1). The average Treg frequency, quantified as the percentage of CD25+ CD127-FoxP3+ cells of total CD4+ cells, was similar at approximately 3% in PBMC and VT, suggesting that a sizeable population of Tregs comparable to blood Treg frequency reside within healthy human vaginal tissue (Fig. 1c). CD69 and integrin αE (CD103) canonically define tissue-resident memory T cells (Trm)33. We quantified CD69 and CD103 expression on Tregs, CD4 Tconv, and CD8 T cells from PBMC and vaginal mucosa. As expected, T cells from blood were largely CD69-CD103−, while a majority (60%) of CD8 cells in the VT were double-positive for CD69 and CD103 (Fig. 1d, e), consistent with our previous report57. A smaller fraction of CD4Tconv (30%) and only 3% of Tregs were CD69+ CD103+, while the majority of vaginal Tregs were CD69+ CD103− (67%), suggesting that CD103 expression may not be necessary for Treg mucosal tissue residency, as has been shown for other CD4+ Trm34,35 (Fig. 1d, e).

a Peripheral blood mononuclear cell (PBMC) and paired mucosal biopsies from the vaginal tract (VT) were obtained from 14 healthy adult women with no genital tract infections. b Tregs were identified by flow cytometry as CD4+ CD127− CD25+ FoxP3+ cells. c Donor-matched frequency of Tregs out of total CD4+ T cells in PBMC and VT. d Representative CD69 and CD103 staining, gated on Tregs (left), and CD69+ and CD103+ fractions of CD8, CD4 Tconv, and Treg populations from PBMC and VT, as a percentage of parent population (right). e CD69+ CD103− (left) and CD69+ CD103+ (right) fractions of CD8, CD4 Tconv, and Tregs from VT. f Representative Treg activation marker staining on PBMC (red) and VT (blue), gated on Tregs (top). Donor-matched frequencies of activation markers in PBMC and VT, gated on Tregs (bottom). n = 14, Significance defined by paired t test; p < 0.05.
We next sought to further phenotype these tissue Tregs. We performed surface and intracellular flow cytometry staining for markers of Treg activation and suppressive capacity, including glucocorticoid-induced tumor necrosis factor-related receptor (GITR), T-cell immunoglobulin and ITIM domain (TIGIT), Cytotoxic T-Lymphocyte Associated Protein 4 (CTLA-4), Inducible T-cell costimulator (ICOS), and CD3958. We found that a significantly higher percentage of VT Tregs compared to blood Tregs expressed each of these markers (mean: 20% vs 4% for ICOS, 85% vs 57% for TIGIT, 76% vs 44% for CD39, 71% vs 22% for CTLA-4, 32% vs 0.4% for GITR, respectively), demonstrating that human vaginal tissue Tregs, at baseline, appear more activated than their circulating counterparts (Fig. 1f). Together, these findings suggest that a pool of highly activated tissue-localized Tregs are maintained in the vaginal mucosa of healthy women and are poised to rapidly modulate immune responses. However, although the biopsies were collected from women without any active infections, we could not rule out the possibility that local inflammation due to environmental antigens or sexual activity was driving activation in vaginal Tregs. To determine if the distinct VT Treg phenotype resulted from vaginal tissue residency alone, or alternatively, arose in response to active inflammatory signals, we employed a mouse model wherein inflammation and antigen exposure could be tightly controlled.
Vaginal tissue Tregs are activated compared to lymphoid Tregs in healthy mice
To characterize VT Tregs, naïve C57BL/6 J mice were synchronized in the diestrus phase of the estrus cycle with a subcutaneous (s.c.) injection of medroxyprogesterone acetate (depo provera) 5–7 days before harvest59. VT and vaginal-draining lymph nodes (dLN) were harvested and stained with fluorescent antibodies for flow cytometry analysis. To exclude T cells derived from the circulation that could be contaminating VT tissues, mice were injected with intravascular (i.v.) CD45.2 antibody 3 min prior to sacrifice60,61 (Supplementary Fig. 2 and Supplementary Table 2) and i.v. label+ cells were excluded from flow cytometry analysis in VT samples. Tregs, identified as CD4+ FoxP3+, were assessed by flow cytometry and quantified as a percentage of total CD4+ T cells in each tissue (Fig. 2a, Supplementary Fig. 2). As previously reported50,51, Tregs in the dLN accounted for approximately 11% of total CD4+ cells; however, we found that they constituted an average of 21% of total vaginal CD4+ cells, demonstrating that Tregs make up a disproportionately large, albeit not statistically significantly different, fraction of vaginal CD4+ T cells (Fig. 2a). Similar to our analysis of human Tregs, we then phenotyped VT and dLN Tregs and found that a significantly higher frequency of VT compared to dLN Tregs expressed activation markers CTLA-4 and GITR at steady state (82% vs 38 % for CTLA-4, 85% vs 56% for GITR) and although not significant, ICOS trended similarly (50% vs 30%; Fig. 2b). However, CD69, which is also an indicator of recent activation62 in addition to being a tissue residency marker, was comparably expressed in VT and dLN Tregs (20% vs 15%; Fig. 2b). These findings demonstrate that Tregs constitute a large percentage of vaginal CD4+ T cells, suggesting that unusually high Treg:Tconv ratios are necessary in the tissue at homeostasis. Thus, the VT Treg phenotype in naïve mice mirrored the heightened activation observed in healthy human VT Tregs, suggesting that VT tissue localization alone is at least partially responsible for the elevated activation and suppressive potential.

C57BL/6 J mice were administered Depo provera s.c. in the neck ruff, and VT and vaginal-draining lymph nodes (dLN) were harvested 5–7 days later. a Tregs were identified as CD4+ FoxP3+ by flow cytometry. b Representative staining for CTLA-4, GITR, ICOS, and CD69 on FoxP3+ Tregs (left; VT plot is the concatenated image of 5 individual VT tissues), quantified as a percentage of total FoxP3+ Tregs (right). n = 5; data representative of 3 replicate experiments. Significance defined by paired t test; p < 0.05.
Vaginal HSV-2 infection increases the accumulation of highly activated Tregs at the site of infection and drives increased expression of select activation markers consistent with a tissue signature
Our lab and others have previously used a mouse model of vaginal herpes simplex virus 2 (HSV-2) infection. To characterize murine VT Treg dynamics upon mucosal infection, mice were injected s.c. with depo provera 5–7 days before intravaginal infection to synchronize estrus cycle phases for consistent susceptibility to HSV-2 infection59. On days 3 and 7 post-infection (p.i.), frequency and phenotypes of dLN and VT Tregs were assessed by flow cytometry. VT Treg frequencies at day 3 p.i. (21%) were stable compared to Treg frequencies at day 0 (21%), but absolute numbers of Tregs were dynamic, significantly increasing by day 7 (mean VT Treg numbers at day 0, 3, and 7, respectively: 128, 263, and 1295) (Fig. 3a). Despite the increase in number of Tregs in the VT, by day 7, Treg frequency decreased to 11%, due to the influx of CD4 effector cells50,51 (Fig. 3a). Conversely, dLN Treg number increased insignificantly p.i. (mean dLN Treg numbers at day 0, 3, and 7, respectively: 150,069, 269,443, 302,729) and their frequency as a percentage of total CD4+ T cells remained stable (11%) out to day 7 (Fig. 3a). These results demonstrate the dynamic nature of Treg in the VT following viral infection, suggesting a Treg role in the early mucosal antiviral responses.

C57BL/6 J mice were administered Depo provera s.c. in the neck ruff, and infected 5-7 days later with intravaginal (ivag.) HSV-2. On days 0 (uninfected [uninf.]), 3, and 7 post-infection, VT and dLN were harvested for flow cytometry analysis. a Tregs were identified as CD4+FoxP3+ by flow cytometry. Representative Treg staining in dLN (grey) and VT (blue) on day 3 post-infection (left). Treg frequency was quantified as a percentage of total CD4+ T cells and absolute number of Tregs on days 0, 3, and 7 (right). b Representative staining for CTLA-4, GITR, ICOS, and CD69 on FoxP3+ Tregs on day 7 post-infection (left), quantified as a percentage of total FoxP3+ Tregs and absolute number on days 0, 3, and 7 (right). n = 5; data representative of 3 replicate experiments. Significance defined by paired t test or Kruskal–Wallis test followed by Dunn’s multiple comparison test; p < 0.05.
We next assessed the Treg phenotype at day 3 and 7 p.i., and found that the frequency of Tregs expressing CTLA-4 and GITR increased only a small amount compared to uninfected mice, as over 80% of Tregs in the uninfected VT were already CTLA-4+ and GITR+ at day 0 (Fig. 3b). However, the absolute numbers of VT Tregs expressing CTLA-4 and GITR increased by day 7 (mean CTLA-4+ Treg number: 1170; GITR: 1193) compared to uninfected mice (mean CTLA-4+ Treg number: 108; GITR: 109; Fig. 3b). Conversely, dLN CTLA-4+ and GITR+ Treg numbers remained stable, and a significantly higher fraction of VT Tregs expressed CTLA-4 and GITR compared to dLN Tregs at both day 3 (CTLA-4: 81% vs 32%; GITR: 82% vs 58%) and day 7 pi (CTLA-4: 89% vs 42%; GITR: 90% vs 57%; Fig. 3b). This suggests that although CTLA-4 and GITR are constitutively expressed on nearly all VT but not dLN Tregs, infection with HSV-2 increases the numbers of these highly activated Tregs in the tissue, and a higher frequency in the VT compared to dLN Tregs consistently express CTLA-4 and GITR after infection.
Notably, the frequency of VT Tregs expressing ICOS was significantly increased at day 7 (80%) compared to uninfected (49%) or day 3 p.i. (40%; Fig. 3b), and the number of VT Tregs expressing ICOS increased significantly by day 7 compared to uninfected (1097 vs 66; Fig. 3b). A higher frequency of VT Tregs expressed ICOS compared to dLN Tregs at both day 3 (41% vs 24%) and day 7 (80% vs 34%; Fig. 3b), suggesting that HSV-2 infection drives potent ICOS expression in local Tregs. We also noted that the frequency of CD69+ VT Tregs (40%) is significantly elevated compared to dLN Tregs (11%) at day 3 p.i. (Fig. 3b). This result highlights the challenge of interpreting CD69 expression, which can be a marker of either tissue residency or activation, outside a well-defined infection model. Together, these findings demonstrate that few Treg are present in the VT tissue of healthy mice, suggesting that an initial inflammatory event or antigenic stimulation is needed to recruit CD4+ T cells to the tissue, but that VT residency alone drives a constitutive, activated Treg phenotype compared to dLN Tregs. Furthermore, viral infection exacerbates the activation phenotype of VT tissue but not dLN Tregs and significantly increases Treg numbers in the VT.
The vaginal Treg population is transcriptionally distinct from lymphoid Tregs
We next sought to further interrogate VT and dLN Tregs at the population level through transcriptional analysis. We infected FoxP3GFP mice63 intravaginally with HSV-2 and harvested VT and dLN on day 7 p.i. We then sorted Tregs (CD4+ GFP+) from VT and dLN from four infected and three uninfected mice and performed bulk RNA sequencing. Uninfected mice contained very few T cells in the VT (Fig. 3a); therefore, we did not recover sufficient Tregs from uninfected VT to obtain reliable sequencing results and this population was excluded from downstream analysis. When comparing Tregs from the VT and dLN of infected mice, we found differential expression of over 30 genes based on tissue location (Fig. 4a). In VT Tregs, the transcripts for canonical Treg genes such as IL-10 were elevated, as was transcription factor Tbx21 (Tbet). Tbet+ Tregs represent a subset of Tregs that are known to be highly suppressive and selectively inhibit T helper type 1 (Th1) responses and CD8+ T cells64. Additionally, transcripts for Ccr2, a tissue-homing chemotactic receptor found on other non-lymhoid tissue Tregs that increases Treg CD25 expression in VAT Tregs, was significantly increased in VT Tregs. We also compared the transcriptional profile of Tregs from infected mice to the transcriptional program of Tregs resident in VAT and found that VT, but not dLN Treg signatures were enriched for genes in a previously published VAT Treg signature, suggesting overlap in the transcriptional profile and functions between non-lymphoid tissue Tregs9 (Fig. 4b). We also identified gene ontology (GO) terms that appear densely in VT Tregs compared to dLN Tregs from infected mice, and found that processes such as cell signaling, chemotaxis, migration, and inflammatory response are enriched in VT Tregs compared to dLN after infection (Supplementary Table 3). Together, our bulk RNA-seq results further confirmed the VT Treg compartment as a distinct population, with both phenotypic and transcriptional differences, compared to lymphoid Tregs.

FoxP3GFP mice were administered Depo provera s.c. in the neck ruff and infected ivag 5–7 days later with WT HSV-2. On day 7 post-infection, VT and dLN were harvested and prepared for fluorescence-activated cell sorting (FACS) to isolate CD4+ FoxP3+ Tregs. Bulk RNA-sequencing was performed on Tregs from dLN and VT. a Significantly differentially expressed genes (DEG) in dLN and VT Tregs were defined by a log2FC greater than 1 and a false discovery rate (FDR) < 0.05 and visualized in a heatmap. n = 4. b Gene set enrichment analysis (GSEA) of dLN and VT Tregs day 7 post-infection compared to a previously published gene signature for visceral adipose tissue Tregs9. n = 4; DEG defined by FDR < 0.05.
Thus far, our data demonstrated that VT tissue Tregs are transcriptionally and phenotypically distinct from dLN Tregs, and that phenotype is further exacerbated after HSV-2 infection, although it was generally difficult to distinguish changes in VT tissue Tregs after infection compared to steady state. Thus, we further explored VT Tregs transcriptionally on a single-cell level. We sorted CD4+ GFP+ Tregs from the VT or dLN of FoxP3GFP mice on day 7 post-HSV-2 infection, or from uninfected control mice. We then performed single-cell RNA-seq (Fig. 5). We used the Seurat pipeline65 to perform graph-based clustering of Tregs in the VT and dLN, visualized with Uniform Manifold Approximation Projection (UMAP)66. As with our bulk RNA-seq, we were unable to recover sufficient VT Tregs from uninfected mice for sequencing. However, we found that VT Tregs from infected mice (shown in blue) grouped largely independently, with some overlap with dLN Tregs from infected mice (shown in grey) and little overlap with Tregs from uninfected dLN (green; Fig. 5a). When we compared differentially expressed genes (DEG) using Model-based Analysis of Single-cell Transcriptomics (MAST)67 in VT Tregs compared with dLN (Fig. 5b), visualized in a heatmap with the top 20 DEG shown for each population, VT Tregs were enriched for genes related to Treg suppression and activation. For example, Tnfrsf18 (GITR), Tigit, and Ctla4 were differentially expressed in VT Tregs compared to dLN Tregs from infected or uninfected mice, as were tissue-homing chemokine receptors Cxcr3 and Ccr2, as well as growth factor Areg, which other mucosal Tregs produce to mediate tissue repair6,7,27 (Supplementary Table 4). Notably, we previously observed by flow cytometry that GITR, TIGIT, and CTLA-4 were increased on vaginal Tregs compared to PBMC or dLN in humans and mice (Figs. 1–3), supporting our findings at the protein level.

FoxP3GFP mice were administered Depo provera s.c. and infected 5–7 days later ivag with WT HSV-2; controls received Depo provera only. On day 7 post-infection, VT and dLN were harvested and prepared for FACS to isolate CD4+ FoxP3+ Tregs. Single-cell RNA-sequencing was performed on dLN and VT Tregs and analyzed with the Seurat pipeline in R. n = 5 HSV-2+ and n = 3 uninfected. a UMAP visualization of graph-based clustering of VT and dLN Tregs from infected mice and dLN Tregs. b Heatmap showing the top 20 DEG in VT and dLN Tregs. Significant DEG were determined by log2FC > 0.5 and FDR < 0.01. A full DEG list can be found in Supplementary Table 4. c Violin plots of GzmB and Nkg7 expression in Treg populations. d, e C57BL/6 J mice were administered Depo provera s.c. in the neck ruff and infected ivag 5–7 days later with HSV-2. On days 0 (uninf.), 3, and 7 post-infection, dLN and VT were harvested for flow cytometry analysis. Tregs and CD8 T cells were identified as CD4+ FoxP3+ and CD8+, respectively. Representative granzyme B (GzmB) ex vivo staining in dLN (grey) and VT (blue) in d Tregs and e CD8 on days 0 and 7 post infection (left), quantified as a percentage of total Treg or CD8+ and by absolute number (right). n = 5 HSV-2+ and n = 5 uninfected. Data representative of three replicate experiments. Significance defined by paired t test or Kruskal–Wallis test followed by Dunn’s multiple comparison test; p < 0.05.
In addition to these genes known to be important for tissue Treg function, we were surprised to find that Gzmb was the most highly upregulated gene in VT compared to dLN Tregs (Fig. 5b, c). This trend was mirrored by Nkg7, a regulator of cytotoxic granule exocytosis which was also increased in VT Tregs by bulk RNA-seq68,69 (Figs. 4a, 5b). GzmB is a serine protease that induces apoptosis through cleavage of caspase substrates in the cytoplasm70. Although more commonly secreted by CD8+ cytotoxic lymphocytes (CTL) and natural killer (NK) cells to kill tumor cells or infected cells, Gzmb+ Tregs, particularly those in mucosal tissue sites such as lung, have been shown to limit effector responses during infection-mediated inflammation, limiting collateral tissue damage29,71,72.
Finally, we also interrogated the transcriptional heterogeneity within dLN and VT Treg populations. Using Seurat and MAST as described above, we performed graph-based clustering of dLN Tregs (HSV-2+ and uninfected) and VT Tregs (HSV-2+), combined. We visualized this clustering with UMAP (Supplementary Fig. 3A) and a heatmap displaying the top 20 DEG per cluster (Supplementary Fig. 3B; a full DEG list can be found in Supplementary Table 5). We found that vaginal Tregs were most highly represented in Cluster 2 (41% of the sequenced VT Treg population; 109 VT-derived Treg cells), and Cluster 2 was comprised of 33% VT-derived Tregs (Supplementary Fig. 3C–E). The DEG enriched in Cluster 2 highly mirrored the DEG enrichments in the infected vaginal Treg compared to infected dLN Treg population described in Fig. 5 and Supplementary Table 4, with genes such as GzmB, Ccr2, Cxcr3, and Nkg7 highly enriched (Supplementary Table 5; Supplementary Fig. 3B). This suggests that the transcriptional differences between VT and dLN Tregs after infection are driven by a large subset of vaginal Tregs. Additionally, Cluster 2 was 59% comprised of Tregs derived from HSV-2-infected dLN (Supplementary Fig. 3D), suggesting that the subset of VT Tregs overlap transcriptionally with a subset of dLN Tregs from infected mice, implicating some shared transcriptional changes that are acquired after infection, regardless of Treg location. Additionally, 19% of VT-derived Tregs grouped within Cluster 0, along with 78% of sequenced dLN Tregs from uninfected mice and a smaller proportion of dLN Tregs from infected mice (Supplementary Fig. 3C, E). Notably, this cluster was defined by activation markers such as Cd8373, Icos, and Tigit, as well as Areg, known for roles in Treg-mediated tissue repair (Supplementary Fig. 3B; Supplementary Table 5). VT-derived Tregs from infected mice also fell within Cluster 1 and to a lesser degree Cluster 3 (Supplementary Fig. 3C–E), while dLN Tregs from infected mice were most highly represented in Clusters 1 and 0 (Supplementary Fig. 3C, E). Together, these results suggest that despite the general transcriptional differences between dLN and VT Tregs, intra-population heterogeneity exists within each group, with some overlap between tissue location. However, a recovery of relatively low cell numbers after quality control filtering in the VT (264 total cells) precludes extensive subsetting and analysis of VT Treg heterogeneity.
Vaginal Tregs express Granzyme B ex vivo after HSV-2 infection
After finding Gzmb transcript highly differentially expressed in VT Tregs after infection, we used flow cytometry to assess GzmB protein expression on vaginal Tregs and CD8+ T cells. In uninfected mice, neither VT (2.5%) nor dLN (0.06%) Tregs highly expressed GzmB (Fig. 5d). CD8+ T cells, which robustly express GzmB in response to viral infection or immunization36,74,75, also expressed little to no GzmB in healthy mice (VT: 5.6%; dLN: .09%; Fig. 5e). However, by day 7 p.i., 19% of VT Tregs were GzmB+, while only 1% of dLN Tregs were GzmB+ (Fig. 5d). Likewise, by day 7, absolute numbers of VT Tregs expressing GzmB increased significantly compared to uninfected mice, as did absolute numbers of dLN Tregs expressing GzmB (Fig. 5d). These results show that VT Tregs have the ability to produce GzmB in a similar pattern to CD8+ CTLs (Fig. 5e). However, our data show that GzmB production is only induced after local infection and is not a general characteristic of vaginal tissue-resident Tregs.
Inflammatory cytokines induce GzmB expression in Tregs
We next sought to identify the signals that regulate expression of GzmB in VT Tregs upon local mucosal infection. We hypothesized that local inflammatory signals induced by infection act on Tregs to induce GzmB during the peak effector phase of the antiviral immune response to HSV-2. Given the high expression of CD25—the interleukin-2 (IL-2) receptor (IL-2Rα)—on Tregs and their dependency on IL-2 for development and function1,58, we hypothesized that IL-2 together with other inflammatory cytokines could induce GzmB. Along these lines, we hypothesized that Type-1 interferons (IFN-I), interferon-α (IFN-α) and interferon-β (IFN-β), which are critical antiviral cytokines produced early in HSV-2 infection by plamacytoid dendritic cells (pDCs)76,77, might act on Tregs through the IFN α/β receptor (IFNAR) to induce GzmB in infected tissue contexts. To test this, we cultured bulk dLN cells (Fig. 6) or splenocytes (Supplementary Fig. 4) from uninfected WT mice for 24 h with IL-2 alone, IFN-α and IFN-β, or all three cytokines together. At 24 h, dLN Tregs, CD4+ Tconv, and CD8+ T cells were assessed for GzmB expression by flow cytometry. We found that IL-2 in combination with IFN-α and IFN-β induced GzmB expression on an average of 5.9% of cultured Tregs, two-fold higher than the average 2.6% Treg expression of GzmB in response to TCR stimulation. In contrast, less than 1% of Tregs in media alone produced GzmB, and less than 1% of CD4+ Tconv expressed GzmB in response to IL-2 in combination with IFN-I (Fig. 6a). Notably, IFN-α/β alone did not induce significant Treg GzmB expression compared to media controls in dLN, while neither IL-2 nor IFN-α/β induced significant Treg GzmB expression in spleen Tregs, suggesting that dual cytokine signals are necessary for robust Treg expression of GzmB (Fig. 6a).

Bulk dLN cells from a healthy C57BL/6 J mouse were cultured in various cytokine conditions as indicated. a After 24 h, CD4+ FoxP3+ Tregs (top), CD4+ FoxP3− CD4Tconv (middle), and CD8+ T cells (bottom) were stained for GzmB expression by flow cytometry. Example staining (left) Tregs, CD4 Tconv, or CD8 expressing GzmB as a percentage of total FoxP3+, CD4+ FoxP3−, or CD8+, respectively (right). b Bulk dLN cells from a healthy mouse were enriched for T cells using magnetic separation and cultured in various cytokine conditions. After 24 h, CD4+ FoxP3+ Tregs (top), CD4+ FoxP3− CD4Tconv (middle), and CD8+ T cells (bottom) were stained for GzmB expression by flow cytometry. Example staining (left) and Tregs, CD4 Tconv, or CD8 expressing GzmB as a percentage of total FoxP3+, CD4− FoxP3+, or CD8+, respectively (right). Data representative of two replicate experiments. Significance defined by one-way ANOVA followed by Tukey’s test for multiple comparisons; p < 0.05.
Inflammatory cytokines act directly on Tregs to induce GzmB expression
We demonstrated that IL-2, in combination with IFN-I, could induce potent GzmB expression on lymphoid Tregs from naïve mice. However, these experiments were performed with bulk dLN cells and splenocytes, where Tregs were cultured with other immune cells that could aid in Treg activation. To parse whether these cytokines can act directly on Tregs or instead were also activating other immune cells that could then produce additional inflammatory cytokines to secondarily activate GzmB expression in Tregs, we repeated these stimulations in cultures lacking antigen presenting cells (APCs). Thus, we purified dLN (Fig. 6b) or splenic T cells (Supplementary Fig. 4B) from naïve mice using pan-T cell negative selection. Although to a lesser degree than our bulk dLN cultures, a significant percentage of Tregs (2.7%) in the pure T cell cultures exposed to IL-2 and IFN-I produced GzmB compared to Tregs cultured in media (less than 1%; Fig. 6b). In spleen, IL-2 alone induced a very small but significant increase in the frequency of Tregs expressing of GzmB (0.88%) on Tregs compared to media control (Supplementary Fig. 4B), while neither IL-2 nor IFN-I induced significant GzmB expression on dLN-derived Tregs. This suggests that IL-2 and IFN-I work synergistically to directly bind CD25 and IFNAR on Tregs to induce GzmB, but when other immune cells are present, this effect is indirectly augmented, likely through the activation of APCs or other immune cells which in turn act on Tregs to increase GzmB expression.
IL-2 is necessary for robust in vivo Treg GzmB expression after HSV-2 infection
After demonstrating that IL-2 in conjunction with IFN-I induces robust GzmB expression on Tregs in vitro, we tested the requirement of IL-2 after infection with HSV-2 for Treg expression of GzmB in vivo. C57BL/6 J mice were infected intravaginally (ivag) with HSV-2 and administered either anti-IL2 (αIL-2) blocking antibody or PBS via i.v. injection on day 3 p.i. To ensure local delivery of αIL-2 to the vaginal cavity, we also administered either αIL-2 or PBS ivag. on days 3–6 p.i., and tissues were examined on day 7–8 p.i. for flow cytometry analysis of GzmB expression. In the dLN, as expected, few Tregs expressed GzmB at all, and αIL-2 treatment did not significantly alter GzmB expression on Tregs (Fig. 7a). However, in the VT, Treg GzmB expression was decreased two-fold, demonstrating that IL-2 is necessary in vivo for full Treg expression of GzmB (Fig. 7a). VT CD8+ T cell expression of GzmB was not significantly decreased by αIL-2 treatment (Fig. 7b), demonstrating a Treg-specific dependence on IL-2 (and likely other inflammatory cytokines such as IFN-I) for GzmB expression. Together, these results validate our in vitro experiments and demonstrate that IL-2 is necessary in vivo for robust GzmB expression on Tregs after HSV-2 infection.

C57BL/6 J mice were administered Depo provera s.c. in the neck ruff and infected ivag 5–7 days later with HSV-2. On day 3 p.i., mice were administered both intravenous (i.v.) and intravaginal (ivag.) anti-IL-2 antibody (aIL-2) or PBS control, and treated with ivag. aIL-2 or PBS on days 4–6. On day 7 or 8 p.i., dLN and VT were harvested for flow cytometry analysis. Tregs and CD8 T cells were identified as CD4+ FoxP3+ and CD8+, respectively. Representative granzyme B (GzmB) ex vivo staining in dLN (grey) and VT (blue) in a Tregs and b CD8 on day 7 post infection (left), quantified as a percentage of total Treg or CD8+ (right). n = 4 HSV-2 + aIL-2, n = 3 HSV-2 + PBS, n = 2 uninfected + aIL-2, n = 1 uninfected + PBS. Data representative of 2 replicate experiments. Significance defined by one-way ANOVA followed by Tukey’s test for multiple comparisions; p < 0.05.
Discussion
We found that vaginal mucosal Tregs in healthy humans express increased CTLA-4, GITR, and ICOS compared to circulating (blood) Tregs. We similarly observed increased expression of CTLA-4, GITR, and ICOS in VT Tregs compared to dLN Tregs in uninfected mice, suggesting that this activated phenotype is driven by vaginal tissue localization. The differences that we observed between vaginal and dLN Tregs are consistent with existing tissue Treg literature showing discrete tissue sites such as skin, lung, muscle, and VAT host Treg populations with unique, activated phenotypes6,7,8,9,10,11,12,13,14. The increased expression of CTLA-4, GITR, and ICOS has been previously reported for steady-state VAT, lung, and skin Tregs, and like VAT Tregs, VT Tregs had a predominantly CD69+ CD103− phenotype8.
To assess whether this VT Treg phenotype is further altered in the context of a local viral infection, we infected mice intravaginally with HSV-2. We found that the vaginal Treg population further increased expression of CTLA-4, GITR, and ICOS indicating a highly activated phenotype consistent with increased suppressor potential. The VT Treg population significantly expanded by day 7 post-infection, while Treg numbers in the dLN remained stable. Although absolute numbers of VT Tregs increased after infection, the frequency of VT Treg expressing GITR and CTLA-4 at steady state was largely unchanged after infection, while the frequency of VT Tregs expressing CD69 was only transiently increased at day 3 p.i. Together, these findings highlight that distinguishing acutely activated VT Tregs from those that are simply localized in the vaginal mucosa is challenging. This is particularly the case if etiological stimuli are not well defined such as in human mucosal barrier tissues.
We next wanted to determine if there are biomarkers in the mouse model system that are able to identify markers of acute VT Treg activation using an RNAseq approach. Notably, we found that after infection with HSV-2, vaginal Tregs differentially express GzmB at both the transcriptional and protein level, demonstrating an inflammation-dependent Treg phenotype that is not induced by vaginal residency alone. Tregs in mouse have been previously shown to express GzmB in response to TCR stimulation and in specific contexts, including roles in colorectal and other cancers29,72,78,79,80,81,82,83. Related to our findings, Loebbermann et al. have previously shown that in the lung, but not in lung-draining lymph nodes, Tregs express GzmB after RSV infection, and that Tregs help to attenuate lung immunopathology by killing CD8+ T cells through a GzmB− mediated mechanism29. Thus, similar to their role in the lung, vaginal Tregs may degranulate GzmB to cull aggressive tissue T cell responses during the height of the anti-HSV-2 immune response, preventing collateral tissue damage. This putative GzmB+ Treg function warrants further study, as does the relationship between GzmB+ Treg and HSV-2 viral clearance, as Treg restraint of vaginal T cells may prevent mucosal damage at the expense of efficient viral control. Additionally, our detection of Areg in VT Tregs after infection potentially indicates an ability to directly contribute to tissue repair through IL-33-mediated Areg production6,7,27. However, elucidation of whether or not VT Tregs can exert repair functions akin to lung and muscle Tregs, either at steady state or following local infection, will require further investigation.
Previous studies have indicated a critical role for TCR signaling to acquire GzmB expression71,79,84. Given the large fraction of GzmB+ VT Tregs, we hypothesized that inflammatory cues could be sufficient to drive GzmB expression85. We found that in combination with IFNα and IFNβ, IL-2 induced GzmB expression in Tregs isolated from both the spleen or dLN of naïve mice. This suggests that the ability of Tregs to produce GzmB is not intrinsic to the Treg itself, but a product of its location—and therefore the cytokine environment—that it experiences. Therefore, we hypothesize that after HSV-2 infection, the local inflammatory cytokine environment leads to upregulation of GzmB on vaginal Tregs. Notably, neither IL-2 nor IFN-I alone induced comparable GzmB expression. This suggests that Tregs require at least two separate signals to acquire GzmB expression, which may prevent aberrant cytotoxicity in inappropriate contexts. Importantly, these cytokines are present in the VT during HSV-2 infection. CD4+ T cells are early responders to local infection, where they produce interferon γ (IFN-γ) and IL-250,51,86,87. Likewise, pDCs produce IFN-I early after infection with HSV-277. We confirmed this hypothesis in vivo, demonstrating that blocking of IL-2 significantly decreases Treg, but not CD8+, expression of GzmB after HSV-2 infection. Thus, our findings demonstrate a mechanism by which vaginal Tregs acquire GzmB during the immune response to HSV-2.
We acknowledge that our study is constrained by low T cell numbers in vaginal tissue, precluding certain analyses. In an effort to compensate for this constraint, we confirmed results in multiple experiments containing large groups of mice and validated our findings at both the transcriptional and protein levels. Of note, our bulk RNAseq analysis did not identify GzmB to be significantly differentially expressed between VT and dLN Treg, though there was a consistent trend of increased expression within the VT, which is in line with our scRNAseq and flow cytometry-based results showing that a fraction, but not all Tregs acquire this phenotype.
We were not able to test the direct function of GzmB+ Tregs, as follow up experiments to address if and how GzmB+ Tregs can kill target cells were not feasible due to the low absolute number of Tregs recovered from murine VT. Given that GzmB acquisition occurs in the absence of agonist TCR signals, potential target cells may be recognized in a TCR-independent manner85,88. Likewise, we recovered insufficient cell numbers to perform informative TCR repertoire analysis from any VT Tregs. Finally, due to the ongoing SARS-CoV-2 pandemic we were unable to obtain additional vaginal biopsies from HSV-2+ women, which could confirm, in parallel with our mouse data, the exacerbated activation phenotype we observed in infected animals compared to uninfected. We hope to interrogate the vaginal immune compartment of HSV-2+ women once recruitment of study participants is again feasible.
In summary, we show that vaginal Tregs are transcriptionally and phenotypically distinct from vaginal-draining lymph node Tregs at steady state. We propose a model wherein a small population of Tregs reside within the vaginal mucosa at homeostasis and are moderately activated compared to lymphoid Tregs. However, vaginal mucosal infection greatly augments vaginal Treg numbers and leads to acquisition of a cytotoxic phenotype, including expression of GzmB in vaginal Tregs, suggesting that viral infection drives a differential phenotype in vaginal Tregs. Additionally, we show that IL-2, in combination with antiviral IFN-I, induce in vitro GzmB expression in Tregs from naïve mice, highlighting a two-signal mechanism by which the acquisition of GzmB is tightly controlled in Tregs, allowing for precise expression during highly inflammatory environments. Our findings demonstrate that the previously uncharacterized vaginal Treg population is dynamic and poised to respond to locally acquired infections with highly suppressive and cytotoxic potential, possibly to restrain aggressive effector T cell responses that may cause collateral tissue damage.
Together, these findings also have implications for mucosal vaccines design. Extensive research has shown that a successful mucosal vaccine must elicit Trm at the site of infection, but so far, these studies have not considered how Tregs may help or hinder local anti-viral T cell responses. Therefore, it is critical to understand how Treg function could be therapeutically leveraged to increase vaccine-elicited antigen-specific Trm responses. Going forward, further work is needed to interrogate the origins, TCR requirement, and functions of vaginal Tregs during HSV-2 and determine how Tregs could be harnessed to support effective viral clearance, Trm responses, and tissue healing.
Experimental procedures
Study population
Healthy, assigned female sex at birth, HIV-1 negative adults (n = 14) were recruited at the Vaccine Trial Unit (Fred Hutchinson Cancer Research Center) in Seattle, WA. Informed consent was obtained from all participants for the collection of blood and vaginal biopsies and was approved by Fred Hutchinson Cancer Research Center internal review board (IR5640). Eligibility criteria included aged ≥18 and <45, non-menopausal, not pregnant, currently using reliable contraceptives, negative for Hepatitis C, HSV-2, and vaginal N. gonorrheae and C. trachomatis and normal PAP smear. A normal vaginal exam at time of biopsy was required. The ages of study participants were 21–47, with a median age of 27. Vaginal biopsies were obtained from the fornices using a Baby Tischler Biopsy Forceps (Wallach Surgical, Trumbull, CT, USA) and lidocaine was not used. All biopsies were transported to the laboratory on wet ice and processed on the day of collection.
Human sample processing
PBMCs were isolated from whole blood by Lymphoprep (Stemcell) gradient and centrifuged for 20 min at 800 × g. Biopsies were prepared for flow cytometry as follows: after mincing, biopsies were digested in collagenase II (700 U/ml; Sigma) and DNAse (400 U/ml; Sigma) at 37 degrees C for 30 min. The digested tissues were passed through a 70 µm strainer to create a single-cell suspension and prepared for flow cytometry analysis.
Mice
6–8 week-old female wildtype C57BL/6 J (Jackson Laboratories, Bar Harbor, ME) or FoxP3GFP 63 (bred at Fred Hutch) mice were used for experimental groups. All animal experiments were approved by Fred Hutch IACUC and the study was conducted in strict compliance with the PHS Policy on Humane Care and Use of Laboratory Animals.
Infections
Mice were injected subcutaneously in the neck ruff with 2 mg of medroxyprogesterone acetate (Depo-Provera) 5–7 days prior to intravaginal (ivag.) infection with 104 PFU of HSV-2 derived from a human clinical isolate.
Intravascular labeling
3 min prior to sacrifice with CO2, mice were administered an intravenous (i.v.) tail vein injection of CD45.2 antibody (3 ug in 200 ul PBS) in PBS to label circulating lymphocytes for exclusion from VT samples.
Mouse tissue processing
Vaginal tract (VT), including the vagina and cervix, was harvested and minced with scissors followed by a 30 min incubation in RPMI containing collagenase D (2 mg/ml; Sigma) and DNAse (15 ug/ml; Sigma) at 37 degrees C. Following incubation, collagenase reaction was quenched with 5 mM EDTA in HBSS without calcium and magnesium and passed through 70 um strainer to prepare a single-cell suspension. After harvesting, vaginal-draining lymph nodes (dLN), including the inguinal and iliac lymph nodes, were harvested and passed through 70 um filter to prepare a single-cell suspension.
Cell sorting and flow cytometry
Cells were incubated in fixable viability dye (Invitrogen) and blocked for non-specific Fc binding and then stained for surface proteins, fixed and permeabilized with Foxp3 Transcription Factor Staining Buffer (eBioscience), and stained for intracellular markers. Cells were collected with the FACSymphony (BD Biosciences, San Jose, CA) and data analyzed with FlowJo software (Treestar, Ashland, OR). For this list of all antibodies used, reference Supplementary Tables 1 and 2.
Cell sorting for RNA-sequencing experiments was performed on the FACSAria (BD Biosciences, San Jose, CA). Tregs and CD4+ Tconv isolated from vaginal tissue and vaginal-draining lymph nodes were sorted from FoxP3GFP mice on CD4+ GFP+ and CD4+ GFP−, respectively. Tregs and CD4+ Tconv from vaginal tissues were additionally sorted on intravascular label- population to exclude blood T cell contamination. To estimate total cell counts, 2 × 104 AccuCheck Counting Beads (Thermo Fisher Scientific) were added to each sample prior to acquisition on the cytometer.
In vitro T cell stimulation assays
For T cell stimulation assays, spleen or dLN were harvested from uninfected C57BL/6 J mice. Pan-T cells were enriched with magnetic T cell negative selection kit (Stem Cell Technologies, Vancouver, Canada) from healthy mouse splenocytes. Single cell suspensions from spleen were either lysed for red blood cells and plated whole or enriched for T cells as described above; dLN cells were plated whole or enriched for T cells as described above. Bulk cells or purified T cells were then cultured at 1e6 cells/well in 200 µl in 96-well plates. Both groups were cultured for 24 h in the presence of recombinant IFNα (Pacific BioLabs; 50 U/ml), recombinant IFNβ (Pacific BioLabs; 50 U/ml), and recombinant IL-2 (Biolegend; 20 ng/ml). Golgiplug (BD Biosciences) was added at 1:1000 after 20 h of culture. For TCR stimulation positive control wells, 96-well plates were coated with 1 ug/ml purified anti-CD3 and 2 ug/ml anti-CD28 (Biolegend) for 24 h at 4 degrees C before plating cells. Negative controls were cultured in media alone.
In vivo IL-2 blocking
On day 3 p.i. with HSV-2, mice were administered 150 ug intravascular anti-IL-2 (aIL-2) antibody (Bio X Cell, clone JES6-1A12) or PBS control. On the same day, mice were also ivag. administered 75 ug anti-IL-2 or PBS control. On days 4–6, mice were administered ivag. 35 ug anti-IL-2 or PBS. On day 7 or 8 p.i., dLN and VT were harvested for flow cytometry analysis.
Statistical analysis
For flow cytometry data, statistical analyses were performed with Prism software (GraphPad Software, San Diego, CA). Paired data were assessed with paired t tests. When comparing groups within datasets containing greater than two groups, Kruskal–Wallis followed by Dunn’s test for multiple comparisions or one-way ANOVA followed by Tukey’s test was used. Significance was defined by a p value less than 0.05.
Bulk RNA-sequencing
We performed bulk RNA-sequencing on 110–200 sorted CD4Tconv and Treg from the dLN of infected and uninfected mice and the VT of infected mice. Cells were not pooled between mice. 11 samples were sequenced: four CD4+ FoxP3+ from infected dLN; 4 CD4+ FoxP3+ from infected VT; three CD4+FoxP3+ from uninfected dLN. Briefly, as previously described89, cells were sorted into SMART-seq v4 Ultra Low Input (Takara Bio USA, San Jose, California) lysis buffer and reverse transcription was performed followed by PCR amplification to generate full length amplified cDNA. Sequencing libraries were constructed using the NexteraXT DNA sample preparation kit (Illumina) to generate Illumina-compatible barcoded libraries. Libraries were pooled and quantified using a Qubit® Fluorometer (Life Technologies). Dual-index, single-read sequencing of the pooled libraries was carried out on a HiSeq2500 sequencer (Illumina) with 58-base reads, using HiSeq v4 Cluster and SBS kits (Illumina) with a target depth of 5 million reads per sample. FASTQs were aligned to a mouse reference genome, using STAR v.2.4.2a and gene counts were generated using htseq-count. QC and metrics analysis was performed using the Picard family of tools (v1.134).
Bulk RNA-seq analysis
The RNA-seq data were aligned to the mouse genome (reference sequence mm10) using STAR (v2.4.2a)90 and gene expression quantification was performed using RSEM (v1.2.22)91. Genes with less than seven nonzero read counts were discarded, leaving 12,317 expressed genes for the analysis. Libraries (samples) with less than 2,000,000 reads; 9000 detected genes; and an exon rate <75% were also removed. Twenty-one of the 22 prepared libraries passed these quality criteria.
Raw count data from RSEM was imported into R. The edgeR Bioconductor package was used to calculate normalization factors92, followed by the voom transformation from the limma Bioconductor package for normalization93,94. Statistical analyses were performed with the limma R package. A linear model was fitted to each gene, and empirical Bayes moderated t-statistics were used to assess differences in expression95. We subsetted the whole dataset into one subset corresponding to the cell type Treg & the treatment HSV-2+, and looked at the constrast between vaginal tract and lymph node. Intraclass correlations were estimated to account for measures originating from the same mice. An absolute log2-fold change cutoff of 1 and a false discovery rate (FDR) cutoff of 5% were used to determine differentially expressed genes (DEGs). Gene set enrichment analysis (GSEA) was performed using the R function Camera implemented within the limma R package96. An FDR cut-off of 5% was used to determine significant gene sets.
Gene Ontology analysis
Pathway analysis of vaginal Tregs compared to dLN Tregs from HSV-2+ mice was performed with the Gene Ontology enRIchment anaLysis and visuaLizAtion tool (GOrilla) http://cbl-gorilla.cs.technion.ac.il/. The entire gene list (ranked from most highly to least highly expressed genes, with VT Tregs as the reference population, including FDR > 0.05 and log2fold change less than 1) were processed with GOrilla for Process terms.
Single-cell RNA-sequencing
Sorted cells from dLN and VT were processed using the 10× Genomics Platform with the 5′ Chromium Single Cell V(D)J Enrichment Kit v2 following manufactures instructions. Sorted populations from each mouse (infected: n = 5; uninfected: n = 3) were pooled with the following total cell numbers were loaded into the Chromium Controller: infected dLN CD4+ FoxP3+: 20,000; infected VT CD4+ FoxP3+: 2855; uninfected dLN CD4 + FoxP3 + : 20,000. As per manufacturer’s instructions, the target cell number for each population was calculated as half the number of cells loaded. cDNA was generated within oil emulsion droplets made by the Chromium Controller. cDNA was purified using DynaBeads MyOne Silane magnetic beads (ThermoFisher, #370002D). cDNA amplification was performed by 10 rounds of PCR using Chromium Single Cell 5′ Reagent kit. Amplified cDNA was then purified with SRPIselect magnetic beads (Beckman Coulter). After cDNA was fragmented and size selected, library construction was performed by end repair, A-tailing, adaptor ligation and PCR (12 cycles). Prior to sequencing, quality of libraries was determined by using Aglient 2200 TapeStation with High Sensitivity D5000 Screen Tape (Agilent). Libraries were quatified by digital droplet PCR with Library Quantification Kit for Illumina TruSeq (BioRad, #1863040). Libraries were diluted to 3 nM and paired-end sequencing was performed on a NovaSeq 6000 sequencer (Illumina).
Single-cell RNA-sequencing analysis
As previously described89, sequencing data were processed to per-cell read and unique molecular identifier (UMI) counts with Cell Ranger software (10× Genomics, Pleasanton, CA). The reads were converted from BCL to FASTQ, demultiplexed, filtered, aligned to mouse genome assembly GRCm38 (mm10), collapsed to UMIs, and assigned to barcodes. Default settings in Cell Ranger were used to call cells vs. background. Data from each sample were aggregated without normalizing.
The R package Seurat was used to analyze count data65,97,98,99. Cells with nCount_RNA values less than 400 and greater than 35,000 were removed, as were cells that had fewer than 400 and greater than 5000 genes detected based on natural breaks in data visualization. Cells with greater than 5% mitochondrial reads were also removed. As previously described89, the UMI counts were log transformed and total UMIs and % mitochondrial reads per cells were regressed out. With UMAP, cells were projected on the first 50 principal component axes from 2000 highly variable genes identified using default cutoffs. Seurat objects were created for each sample and re-scaled using all genes. Each cell’s expression of defined gene sets was summarized by the mean normalized expression across the set in each cell. To remove noise around 0 introduced by normalization, 0 values for each gene in each cell with a raw UMI count of 0 were restored. Differential expression was quantified among populations using the hurdle model in MAST, with total UMIs per cell as a covariate. MAST identifies DE genes that overlap with those found by other methods with high precision and low false positive rates67,100.
Data availability
The sequencing data from this publication have been deposited in the NCBI’s Gene Expression Omnibus and are accessible through the series accession number GEO: GSE189375. All scripts used for data processing and figure generation are available at GitHub: https://github.com/Brianna-Traxinger/scRNAseq_vaginalTreg_HSV-2.
References
Fontenot, J. D., Gavin, M. A. & Rudensky, A. Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 4, 330–336 (2003).
Hori, S., Nomura, T. & Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061 (2003).
Josefowicz, S. Z. & Rudensky, A. Control of regulatory T cell lineage commitment and maintenance. Immunity 30, 616–625 (2009).
Khattri, R., Cox, T., Yasayko, S. A. & Ramsdell, F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 4, 337–342 (2003).
Williams, L. M. & Rudensky, A. Y. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat. Immunol. 8, 277–284 (2007).
Kuswanto, W. et al. Poor repair of skeletal muscle in aging mice reflects a defect in local, interleukin-33-dependent accumulation of regulatory T cells. Immunity 44, 355–367 (2016).
Burzyn, D. et al. A special population of regulatory T cells potentiates muscle repair. Cell 155, 1282–1295 (2013).
Feuerer, M. et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 15, 930–939 (2009).
Cipolletta, D. et al. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 486, 549–553 (2012).
Li, C. et al. TCR transgenic mice reveal stepwise, multi-site acquisition of the distinctive Fat-Treg Phenotype. Cell 174, 285–299 e212 (2018).
Kalekar, L. A. et al. Regulatory T cells in skin are uniquely poised to suppress profibrotic immune responses. Sci. Immunol. 4, eaaw2910 (2019).
Ali, N. et al. Regulatory T cells in skin facilitate epithelial stem cell differentiation. Cell 169, 1119–1129 e1111 (2017).
Mathur, A. N. et al. Treg-cell control of a CXCL5-IL-17 inflammatory axis promotes hair-follicle-stem-cell differentiation during skin-barrier repair. Immunity 50, 655–667 e654 (2019).
Sanchez Rodriguez, R. et al. Memory regulatory T cells reside in human skin. J. Clin. Invest. 124, 1027–1036 (2014).
Shafiani, S., Tucker-Heard, G., Kariyone, A., Takatsu, K. & Urdahl, K. B. Pathogen-specific regulatory T cells delay the arrival of effector T cells in the lung during early tuberculosis. J. Exp. Med. 207, 1409–1420 (2010).
Kolodin, D. et al. Antigen- and cytokine-driven accumulation of regulatory T cells in visceral adipose tissue of lean mice. Cell Metab. 21, 543–557 (2015).
Tanoue, T., Atarashi, K. & Honda, K. Development and maintenance of intestinal regulatory T cells. Nat. Rev. Immunol. 16, 295–309 (2016).
Cosovanu, C. & Neumann, C. The many functions of Foxp3(+) regulatory T cells in the intestine. Front Immunol. 11, 600973 (2020).
Noval Rivas, M. & Chatila, T. A. Regulatory T cells in allergic diseases. J. Allergy Clin. Immunol. 138, 639–652 (2016).
Singh, R. et al. Regulatory T cells in respiratory health and diseases. Pulm. Med. 2019, 1907807 (2019).
Chatila, T. A. et al. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J. Clin. Invest. 106, R75–R81 (2000).
Verbsky, J. W. & Chatila, T. A. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and IPEX-related disorders: an evolving web of heritable autoimmune diseases. Curr. Opin. Pediatr. 25, 708–714 (2013).
Kearley, J., Barker, J. E., Robinson, D. S. & Lloyd, C. M. Resolution of airway inflammation and hyperreactivity after in vivo transfer of CD4+CD25+ regulatory T cells is interleukin 10 dependent. J. Exp. Med. 202, 1539–1547 (2005).
Lewkowich, I. P. et al. CD4+CD25+ T cells protect against experimentally induced asthma and alter pulmonary dendritic cell phenotype and function. J. Exp. Med. 202, 1549–1561 (2005).
Josefowicz, S. Z. et al. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 482, 395–399 (2012).
Hartl, D. et al. Quantitative and functional impairment of pulmonary CD4+CD25hi regulatory T cells in pediatric asthma. J. Allergy Clin. Immunol. 119, 1258–1266 (2007).
Arpaia, N. et al. A distinct function of regulatory T cells in tissue protection. Cell 162, 1078–1089 (2015).
Belkaid, Y., Piccirillo, C. A., Mendez, S., Shevach, E. M. & Sacks, D. L. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature 420, 502–507 (2002).
Loebbermann, J. et al. Regulatory T cells expressing granzyme B play a critical role in controlling lung inflammation during acute viral infection. Mucosal Immunol. 5, 161–172 (2012).
Mendez, S., Reckling, S. K., Piccirillo, C. A., Sacks, D. & Belkaid, Y. Role for CD4(+) CD25(+) regulatory T cells in reactivation of persistent leishmaniasis and control of concomitant immunity. J. Exp. Med. 200, 201–210 (2004).
Shafiani, S. et al. Pathogen-specific Treg cells expand early during mycobacterium tuberculosis infection but are later eliminated in response to Interleukin-12. Immunity 38, 1261–1270 (2013).
Suvas, S., Azkur, A. K., Kim, B. S., Kumaraguru, U. & Rouse, B. T. CD4+CD25+ regulatory T cells control the severity of viral immunoinflammatory lesions. J. Immunol. 172, 4123–4132 (2004).
Gebhardt, T. et al. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 10, 524–530 (2009).
Turner, D. L. & Farber, D. L. Mucosal resident memory CD4 T cells in protection and immunopathology. Front Immunol. 5, 331 (2014).
Woodward Davis, A. S. et al. The human memory T cell compartment changes across tissues of the female reproductive tract. Mucosal Immunol. 14, 862–872 (2021).
Dave, V. A. et al. Cervicovaginal Tissue Residence Confers a Distinct Differentiation Program upon Memory CD8 T Cells. J. Immunol. 206, 2937–2948 (2021).
Iijima, N. & Iwasaki, A. T cell memory. A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science 346, 93–98 (2014).
Roychoudhury, P. et al. Tissue-resident T cell-derived cytokines eliminate herpes simplex virus-2-infected cells. J. Clin. Invest. 130, 2903–2919 (2020).
Schiffer, J. T. Mucosal HSV-2 specific CD8+ T-cells represent containment of prior viral shedding rather than a correlate of future protection. Front Immunol. 4, 209 (2013).
Shin, H. & Iwasaki, A. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature 491, 463–467 (2012).
Dropulic, L. K. & Cohen, J. I. The challenge of developing a herpes simplex virus 2 vaccine. Expert Rev. Vaccines 11, 1429–1440 (2012).
Shin, H. & Iwasaki, A. Generating protective immunity against genital herpes. Trends Immunol. 34, 487–494 (2013).
Milman, N. et al. In situ detection of regulatory T cells in human genital herpes simplex virus type 2 (HSV-2) reactivation and their influence on spontaneous HSV-2 reactivation. J. Infect. Dis. 214, 23–31 (2016).
Smigiel, K. S., Srivastava, S., Stolley, J. M. & Campbell, D. J. Regulatory T-cell homeostasis: steady-state maintenance and modulation during inflammation. Immunol. Rev. 259, 40–59 (2014).
Richert-Spuhler, L. E. & Lund, J. M. The immune Fulcrum: regulatory T cells tip the balance between Pro- and Anti-inflammatory outcomes upon infection. Prog. Mol. Biol. Transl. Sci. 136, 217–243 (2015).
Vick, S. C. et al. A regulatory T cell signature distinguishes the immune landscape of COVID-19 patients from those with other respiratory infections. Sci. Adv. 7, eabj0274 (2021).
Brincks, E. L. et al. Antigen-specific memory regulatory CD4+Foxp3+ T cells control memory responses to influenza virus infection. J. Immunol. 190, 3438–3446 (2013).
Botta, D. et al. Dynamic regulation of T follicular regulatory cell responses by interleukin 2 during influenza infection. Nat. Immunol. 18, 1249–1260 (2017).
Leon, B., Bradley, J. E., Lund, F. E., Randall, T. D. & Ballesteros-Tato, A. FoxP3+ regulatory T cells promote influenza-specific Tfh responses by controlling IL-2 availability. Nat. Commun. 5, 3495 (2014).
Lund, J. M., Hsing, L., Pham, T. T. & Rudensky, A. Y. Coordination of early protective immunity to viral infection by regulatory T cells. Science 320, 1220–1224 (2008).
Soerens, A. G., Da Costa, A. & Lund, J. M. Regulatory T cells are essential to promote proper CD4 T-cell priming upon mucosal infection. Mucosal Immunol. 9, 1395–1406 (2016).
Traxinger, B. R., Richert-Spuhler, L. E. & Lund, J. M. Mucosal tissue regulatory T cells are integral in balancing immunity and tolerance at portals of antigen entry. Mucosal Immunol. 15, 398–407 (2022).
Aluvihare, V. R., Kallikourdis, M. & Betz, A. G. Regulatory T cells mediate maternal tolerance to the fetus. Nat. Immunol. 5, 266–271 (2004).
Guerin, L. R. et al. Seminal fluid regulates accumulation of FOXP3+ regulatory T cells in the preimplantation mouse uterus through expanding the FOXP3+ cell pool and CCL19-mediated recruitment. Biol. Reprod. 85, 397–408 (2011).
Moldenhauer, L. M. et al. Cross-presentation of male seminal fluid antigens elicits T cell activation to initiate the female immune response to pregnancy. J. Immunol. 182, 8080–8093 (2009).
Robertson, S. A. et al. Seminal fluid drives expansion of the CD4+CD25+ T regulatory cell pool and induces tolerance to paternal alloantigens in mice. Biol. Reprod. 80, 1036–1045 (2009).
Pattacini, L. et al. A pro-inflammatory CD8+ T-cell subset patrols the cervicovaginal tract. Mucosal Immunol. 12, 1118–1129 (2019).
Vignali, D. A., Collison, L. W. & Workman, C. J. How regulatory T cells work. Nat. Rev. Immunol. 8, 523–532 (2008).
Linehan, M. M. et al. In vivo role of nectin-1 in entry of herpes simplex virus type 1 (HSV-1) and HSV-2 through the vaginal mucosa. J. Virol. 78, 2530–2536 (2004).
Anderson, K. G. et al. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat. Protoc. 9, 209–222 (2014).
Galkina, E. et al. Preferential migration of effector CD8+ T cells into the interstitium of the normal lung. J. Clin. Invest. 115, 3473–3483 (2005).
Han, Y., Guo, Q., Zhang, M., Chen, Z. & Cao, X. CD69+ CD4+ CD25- T cells, a new subset of regulatory T cells, suppress T cell proliferation through membrane-bound TGF-beta 1. J. Immunol. 182, 111–120 (2009).
Fontenot, J. D. et al. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity 22, 329–341 (2005).
Levine, A. G. et al. Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature 546, 421–425 (2017).
Stuart, T. et al. Comprehensive Integration of Single-. Cell Data. Cell 177, 1888–1902 e1821 (2019).
Becht, E. et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 37, 38–44 (2018).
Finak, G. et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 278 (2015).
Malarkannan, S. NKG7 makes a better killer. Nat. Immunol. 21, 1139–1140 (2020).
Ng, S. S. et al. The NK cell granule protein NKG7 regulates cytotoxic granule exocytosis and inflammation. Nat. Immunol. 21, 1205–1218 (2020).
Velotti, F., Barchetta, I., Cimini, F. A. & Cavallo, M. G. Granzyme B in inflammatory diseases: apoptosis, inflammation, extracellular Matrix Remodeling, Epithelial-to-Mesenchymal Transition and Fibrosis. Front. Immunol. 11, 587581 (2020).
Grossman, W. J. et al. Differential expression of granzymes A and B in human cytotoxic lymphocyte subsets and T regulatory cells. Blood 104, 2840–2848 (2004).
Zhao, D. M., Thornton, A. M., DiPaolo, R. J. & Shevach, E. M. Activated CD4+CD25+ T cells selectively kill B lymphocytes. Blood 107, 3925–3932 (2006).
Doebbeler, M. et al. CD83 expression is essential for Treg cell differentiation and stability. JCI Insight 3, e99712 (2018).
Kohlmeier, J. E., Cookenham, T., Roberts, A. D., Miller, S. C. & Woodland, D. L. Type I interferons regulate cytolytic activity of memory CD8(+) T cells in the lung airways during respiratory virus challenge. Immunity 33, 96–105 (2010).
Mackay, L. K. et al. Maintenance of T cell function in the face of chronic antigen stimulation and repeated reactivation for a latent virus infection. J. Immunol. 188, 2173–2178 (2012).
Lund, J., Sato, A., Akira, S., Medzhitov, R. & Iwasaki, A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 198, 513–520 (2003).
Lund, J. M., Linehan, M. M., Iijima, N. & Iwasaki, A. Cutting Edge: Plasmacytoid dendritic cells provide innate immune protection against mucosal viral infection in situ. J. Immunol. 177, 7510–7514 (2006).
Cao, X. et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity 27, 635–646 (2007).
Sula Karreci, E. et al. Human regulatory T cells undergo self-inflicted damage via granzyme pathways upon activation. JCI Insight 2, e91599 (2017).
Sun, B., Liu, M., Cui, M. & Li, T. Granzyme B-expressing treg cells are enriched in colorectal cancer and present the potential to eliminate autologous T conventional cells. Immunol. Lett. 217, 7–14 (2020).
Gondek, D. C., Lu, L. F., Quezada, S. A., Sakaguchi, S. & Noelle, R. J. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J. Immunol. 174, 1783–1786 (2005).
Salti, S. M. et al. Granzyme B regulates antiviral CD8+ T cell responses. J. Immunol. 187, 6301–6309 (2011).
Dolina, J. S. et al. Developmentally distinct CD4(+) Treg lineages shape the CD8(+) T cell response to acute Listeria infection. Proc. Natl Acad. Sci. USA 119, e2113329119 (2022).
Efimova, O. V. & Kelley, T. W. Induction of granzyme B expression in T-cell receptor/CD28-stimulated human regulatory T cells is suppressed by inhibitors of the PI3K-mTOR pathway. BMC Immunol. 10, 59 (2009).
Maurice, N. J., Taber, A. K. & Prlic, M. The Ugly Duckling Turned to Swan: a change in perception of bystander-activated memory CD8 T cells. J. Immunol. 206, 455–462 (2021).
Milligan, G. N. & Bernstein, D. I. Interferon-gamma enhances resolution of herpes simplex virus type 2 infection of the murine genital tract. Virology 229, 259–268 (1997).
Nakanishi, Y., Lu, B., Gerard, C. & Iwasaki, A. CD8(+) T lymphocyte mobilization to virus-infected tissue requires CD4(+) T-cell help. Nature 462, 510–513 (2009).
Chu, T. et al. Bystander-activated memory CD8 T cells control early pathogen load in an innate-like, NKG2D-dependent manner. Cell Rep. 3, 701–708 (2013).
Woodward Davis, A. S. et al. The human tissue-resident CCR5(+) T cell compartment maintains protective and functional properties during inflammation. Sci. Transl. Med. 11, eaaw8718 (2019).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinforma. 12, 323 (2011).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Law, C. W., Chen, Y., Shi, W. & Smyth, G. K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3: Article3 (2004).
Wu, D. & Smyth, G. K. Camera: a competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res. 40, e133 (2012).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529 (2021).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Satija, R., Farrell, J. A., Gennert, D., Schier, A. F. & Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502 (2015).
Wang, T., Li, B., Nelson, C. E. & Nabavi, S. Comparative analysis of differential gene expression analysis tools for single-cell RNA sequencing data. BMC Bioinforma. 20, 40 (2019).
Acknowledgements
We thank the members of the Lund and Prlic labs for their helpful input and discussions and the study participants. Select figure graphics were created with Biorender.com.
Funding
This work was funded by the National Institute of Allergy and Infectious Disease of the US National Institutes of Health (R01 AI141435 and AI131914 to J.M.L.). J.R.E. and S.C.V. were funded by the Diseases of Public Health Importance Training Grant (T32 AI007509) and B.R.T. was funded by the Viral Pathogenesis Training Grant (T32 AI083203).
Author information
Authors and Affiliations
Contributions
B.R.T., S.C.V., A.W.D., and J.R.E. performed all experiments. B.R.T. and V.V. performed data and statistical analyses. J.C. and C.T. collected the human clinical samples. B.R.T., M.P., and J.M.L. designed the study and wrote the first draft of the manuscript. All authors contributed to editing and approved the final draft.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Traxinger, B., Vick, S.C., Woodward-Davis, A. et al. Mucosal viral infection induces a regulatory T cell activation phenotype distinct from tissue residency in mouse and human tissues. Mucosal Immunol 15, 1012–1027 (2022). https://doi.org/10.1038/s41385-022-00542-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-022-00542-7
