
Abstract
Inflammatory bowel disease (IBD) is characterized by a dysregulated intestinal epithelial barrier leading to breach of barrier immunity. Here we identified similar protein expression changes between IBD and Citrobacter rodentium-infected FVB mice with respect to dysregulation of solute transporters as well as components critical for intestinal barrier integrity. We attribute the disease associated changes in the model to the emergence of undifferentiated intermediate intestinal epithelial cells. Prophylactic treatment with IL-22.Fc in C. rodentium-infected FVB mice reduced disease severity and rescued the mice from lethality. Multi-omics and solute analyses revealed that IL-22.Fc treatment prevented disease-associated changes including disruption of the solute transporter machinery and restored proper physiological functions of the intestine, respectively. Taken together, we established the disease relevance of the C. rodentium-induced colitis model to IBD, demonstrated the protective role of IL-22 in amelioration of epithelial dysfunction and elucidated the molecular mechanisms with IL-22’s effect on intestinal epithelial cells.
Similar content being viewed by others

Introduction
Inflammatory Bowel Disease (IBD), comprised of Crohn’s Disease (CD) and Ulcerative Colitis (UC), is a chronic inflammatory disease of the intestine1. The etiology of IBD is complex, and involves an interplay between host genetics, microbial dysbiosis, and a malfunctioning immune system2. One of the key mechanisms underlying IBD is dysregulated intestinal barrier function as well as epithelial tissue damage leading to a breach in barrier immunity that leads to colitis3. Mucosal healing therapies that restore the barrier and epithelial function may result in long-term remission in IBD3,4. There is heterogeneity in intestinal epithelial cells (IECs) in healthy individuals as well as UC patients5. In UC patients, IECs express elevated markers of inflammation, while genes involved in reinforcing barrier integrity, including preventing invasion by bacteria, were reduced. Interestingly, Parikh et al.5 reported an increase in Wnt signaling modulator PLA2G2A and reduction of chloride absorptive transport machinery (CAR1 and SLC26A2) in colonocytes from UC patients; this indicates that UC patient colonocytes are characterized by overt proliferation at the expense of differentiation/function. Thus, preclinical models that mimic IBD epithelial dysfunction are essential to understand the pathophysiology of IBD.
Due to heterogeneity in the mechanisms that drive IBD, several pre-clinical models are used to interrogate the biology of IBD6. One such model that mimics microbial-driven mechanisms is Citrobacter rodentium infectious colitis7. C. rodentium uses similar infection strategies to breach the intestinal barrier as human enteropathogenic Escherichia coli and induces host microbiota dysbiosis, which is thought to drive the pathophysiology of IBD (reviewed in7,8). Infection of mice with C. rodentium induces damage to colonic IECs and eventually leads to epithelial hyperplasia, which results in either self-limiting disease in C57BL/6 or fatal colitis in strains such as FVB9. The loss of disease tolerance in FVB mice is attributed to excessive Wnt-mediated epithelial cell proliferation and dysfunction, and notably dysregulation of solute transporters that control the physiological balance of Na+/Cl−10,11. Hence, the C. rodentium colitis model could be relevant to the study of epithelial dysfunction in IBD.
IL-22 was recently reported to inhibit Wnt signaling in mouse intestinal organoids12, raising the possibility that IL-22 could rescue the Wnt signaling defects in the intestinal epithelium of infected FVB mice. IL-22 plays a critical role in host defense and epithelial cell homeostasis4. IL-22 is secreted by lymphocytes and signals via its receptor, a complex of IL22RA1/IL10RB, which is highly expressed in various IEC lineages13. Pre-clinical studies established the critical role of IL-22 in intestinal tissue repair and homeostasis. Genetic ablation of IL-22 signaling via deficiencies in either IL-22 production or its receptor IL22RA1, as well as neuralization of IL-22, increased susceptibility to dextran sulfate sodium (DSS), T cell transfer (TCT) or C. rodentium induced colitis in C57BL/6 mice14,15. In these models, IL-22 treatment repaired the barrier by inducing epithelial cell proliferation/survival and by reinforcing the barrier via production of mucin from goblet cells (reviewed in4). Moreover, IL-22 potentiates host defense by stimulating anti-microbial peptides (AMPs) and prevents growth of pathogenic microbial species by inducing glycosylation of host cell proteins that act as an energy source for probiotic species16. In line with its multifaceted role, treatment with an IL-22.Fc fusion protein ameliorated DSS colitis17. IL-22.Fc also increased paracellular water transport that facilitated clearance of C. rodentium in infected C57BL/6 mice15. Based on pre-clinical data, treatment with IL-22 is being investigated as a treatment for IBD18,19.
In this study, we first compared the differential expression of proteins critical for IEC function and differentiation from C. rodentium-infected FVB mice and used proteomics analyses in IBD patients to understand the translatability of the model to human disease20. Our proteomics analyses revealed similarities between human IBD and infected FVB mice. Next, we also assessed whether prophylactic treatment with IL-22.Fc would ameliorate disease severity and found that IL-22.Fc treatment rescued infected mice from lethality by preventing fatal diarrhea. IL-22 prevented the disease associated dysregulation of critical solute transporters and the emergence of intermediate intestinal epithelial cells, which overlapped with undifferentiated IECs present in human tissues5. Taken together, these studies elucidated a mouse model with hallmarks of epithelial dysfunction noted in IBD patients and expanded the mechanism of action (MoA) for mucosal healing during therapy with IL-22.Fc.
Results
Proteomics and single cell CITE-seq analyses highlight the parallels and differences between human IBD and C. rodentium induced colitis in a susceptible mouse strain
Multi-omics studies have revealed IEC defects in human IBD5, and revealed the presence of undifferentiated IECs with drastically reduced expression of chloride absorptive transporters (e.g., SLC26A3 and CAR4), and components of the mucous layer (e.g., MUC2 and CEACAM1), which play key roles in regulating intestinal homeostasis. Notably, down-regulation of both Slc26a3 and Car4 has been observed in C. rodentium-infected susceptible mice (such as FVB10) suggesting that this model could be used to study intestinal epithelial dysfunction. To that end and to understand the translatability of this model to human disease, we conducted a proteomics analysis in C. rodentium-infected FVB mice and assessed protein expression changes for key proteins that regulate intestinal homeostasis in the model and compared these results with our previously published IBD proteomics dataset20.
After inoculating FVB mice orally with C. rodentium, we confirmed an increase in fecal CFU on days 5 and 7 post-infection (p.i.) in colon tissue compared to non-infected control mice (Supplementary Fig. 1a). To better understand disease kinetics in infected mice, we established a novel endoscopy disease severity score, i.e., the Murine Endoscopic Disease Activity Index (MEDAI), which was defined based on thickening of the intestinal epithelium, presence of exudate and loss of vascularity (scoring system described in Supplementary Table 1). We observed an increase in the MEDAI as early as day 5 p.i., which peaked on day 7 p.i. and remained severe until day 9 p.i. (Fig. 1a; representative images shown in Supplementary Fig. 1b). Endoscopically, the colitis in C. rodentium-infected FVB mice was characterized by a white intestinal surface due to hyperplasia-induced increased granularity and release of exudate resulting in masking of vascularity (Supplementary Fig. 1b). By day 7 p.i, epithelial hyperproliferation led to angular protrusions of the thickened epithelium into the lumen resulting in a striated appearance, a hallmark of disease progression in this model (Supplementary Fig. 1b). Consistent with published literature21, we observed that 90% of the FVB mice exhibited severe clinical disease as manifested by mice that succumbed to infection or were euthanized after the loss of >20% body weight/moribund appearance by day 10 p.i. (Supplementary Fig. 1c). Disease kinetics reported by endoscopy were confirmed by histological analyses and Alcian blue staining of goblet cells, which showed progressive inflammation, mucosal hyperplasia and goblet cell loss in the distal colon (Supplementary Fig. 1d–1f). We observed that some crypts in CD and UC tissues, but not all, showed the loss of goblet cells, which is a sporadic feature that occurred in certain areas of the tissue (Supplementary Fig. 1g; indicated by the arrow), consistent with previous reports22,23,24.

a FVB mice were infected with C. rodentium p.o. and disease progression was measured using endoscopy at various times p.i. (n = 5–9 mice per time point). b Volcano plot depicting protein expression changes on day 8 p.i. compared to naïve mice detected using MS based proteomics. c Heat map of select protein expression changes in infected FVB mice with previously published CD or UC proteomics datasets20, compared to respective healthy controls. d IPA pathway, upstream regulator and function comparisons between mouse cell Cluster 4 and a human highly activated T cell cluster. e IPA pathway, upstream regulator and function comparisons between mouse cell Cluster 3 and a human inflammatory macrophage cluster. *p < 0.05, ***p < 0.001 determined as follows: Kruskal–Wallis with Dunnett’s multiple comparisons test (a). Data are representative of five independent experiments (a). Horizontal bars represent the mean of the points shown (a).
We performed a Mass-Spectrometry (MS) based global proteomics analysis of mouse colon tissues on day 8 p.i., at the peak of MEDAI, from naïve mice and untreated infected mice. As expected, a heat map showed that infection-induced major alterations in protein expression (Supplementary Fig. 2a). A volcano plot revealed that CAR4 and SLC26A3 were significantly downregulated compared with naïve controls (Fig. 1b), consistent with published data10. In diseased untreated mice, we observed a strong induction of the pro-proliferative/tumorigenic cycle regulator CDC20 and the Wnt signaling modulator PLA2G2A25. PLA2GA is expressed by secretory cells in the mouse colonic tissue and its secreted form, induced upon inflammation, drives proliferation of IECs in FVB mice25. Interestingly, PLA2G2A mRNA is increased in UC patient colonocytes compared to controls5. In our study, PLA2G2A protein level was not altered in human diseased tissues (data not shown). We also observed down-regulation of the SULT1 family of proteins and HAO2, which have been observed in other models of colitis, thus highlighting similarities between pre-clinical models of disease26.
Among the proteins critical for intestinal homeostasis, we analyzed changes in chloride absorptive transporters (SLC26A3 and CAR 1/2/4), a chloride secretory transporter (SLC12A2), components of the Na+/K+-ATPase (ATP1B1/B2) that establish electrochemical gradients critical for movement of Na+, K+ and Cl−, mucus components (MUC2, KLK1, CEACAM1, LGALS3, DMBT1, PIGR), a regulator of goblet cell differentiation (AGR2), differentiation markers for IECs (KRT19, S100A6, SELENBP1) and proteins involved in preventing bacterial invasion (DMBT1 and ZG16)5,27,28,29. Comparing mouse proteomics data to our published proteomics analyses in human IBD, we observed down-regulation of chloride absorptive machinery during disease in mice and humans, compared to healthy controls (Fig. 1c). In the same way, some components of the mucous layer i.e. MUC2, KLK1, LGALS3, DMBT1 and ZG16, also shared similar protein expression changes in infected mice and IBD tissues (Fig. 1c).
We examined expression of tight and adherens junction proteins that are parts of the IEC layer30. By proteomics, we found there was modest change in CLDN3, CDH1, CDH17 and TJP1-3 after infection (Fig. 1b) whereas they were more prominent changes in IBD patients (Fig. 1c). We also analyzed changes in proteins in involved wound repair31 and found that the extent of the protein changes was modest in both the mouse disease and human IBD (Supplementary Fig. 2b and Fig. 1c). While differences in fold expression between mouse and human diseased tissues as well as between UC and CD tissues were observed, we found that our proteomics analyses identified similar patterns of dysregulation of key intestinal homeostatic proteins involved in solute transport and mucous layer in C. rodentium-induced colitis in FVB mice and in human IBD.
We also assessed changes in disease relevant immune cells such as T cells and macrophages at the same time point when the IECs were assessed, to address whether they undergo similar changes in C. rodentium-induced colitis as human IBD. We performed cell type label transfer from CD patients32 to mouse cell clusters based on transcriptomic signatures and found that a majority of Cluster 4 cells are highly activated T cells in CD patients and that Cluster 3 cells were inflammatory macrophages in CD (Supplementary Tables 2 and 3). We then compared gene expression changes after C. rodentium infection in mouse T cells (Cluster 4) and macrophages (Cluster 3) to their counterpart cell populations in CD. IPA analysis revealed that several pathways were regulated similarly between mouse T cells after C. rodentium infection and human highly activated T cells from IBD patients (Fig. 1d). Such pathways included leukocyte extravasation, T cell receptor activation, and Th1 and Th17 responses (Fig. 1d). Mouse and human T cell changes shared similar upstream regulators and cellular functions such as cell migration, differentiation, and proliferation (Fig. 1d). In contrast to T cells, mouse and human macrophages had less similarities based on IPA pathway analyses, although mouse and human macrophages performed similar cellular functions as evidenced by shared function terms including cell migration (Fig. 1e). Future studies will address the alterations in immune cells that occur during the early phases and the impact of IL-22 treatment.
IL-22.Fc prevented colitis and improved survival in C. rodentium-infected FVB mice
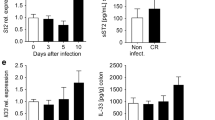
Several studies indicated that fatal colitis in FVB mice is due to infection-induced epithelial hyperplasia potentially due to an increase in Wnt signaling modulators, including RSPO2 and/or secreted PLA2G2A11,25. IL-22 inhibits Wnt and NOTCH signaling in mouse intestinal organoid cultures12. Moreover, due to the multi-faceted role of IL-22 in regulating several other aspects of intestinal epithelium homeostasis (reviewed in4,18), we assessed the secretion of IL-22 from colonic explants following C. rodentium-infected mice. We found that IL-22 secretion peaked on day 3 p.i. and decreased to basal level on day 7 and 9 p.i. from C. rodentium-infected mice when compared to naïve mice (Fig. 2a), consistent with a previous study33. The reduced levels of IL-22 C. rodentium-infected FVB on day 7 and 9 p.i. could be the cause of disease worsening in the infected FVB mice. This prompted us to determine whether exogenous IL-22.Fc would ameliorate disease in infected FVB mice and rescue them from lethality.

Prophylactic treatment of IL-22.Fc was started before infection and administered twice per week until the end of the experiment (n = 3–10 mice per group). a IL-22 secretion from colonic explants of naïve non-infected mice and C. rodentium infected FVB mice at various times p.i. b Schematic of prophylactic treatment of IL-22.Fc. On day 5 p.i., fecal burdens (c), and on day 7 p.i. endoscopic disease severity (d), distal colonic inflammation (left) and hyperplasia (right) in H&E stained tissues (e) were measured. f Representative endoscopy (top) and H&E staining (bottom) images of distal colon in various treatment groups. Scale bar, 50 μm. g Percent survival curves from the start of the experiment until 90% of the untreated animals were either found dead or exhibited >20% body weight lost. h–j Immunoblot analyses of REG3B, REG3G, PHGR1 and GAPDH (loading control) in various treatment groups (data from individual mice shown in h and i) and densitometric quantification (j). NS, not statistically significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 determined as follows: One sample Wilcoxon test or Mann–Whitney test multiplied by the number of inter-group comparisons (c–e), log-rank test (g), or one-way ANOVA with Dunnett’s multiple comparisons test (j). Data are representative of two (a) or four independent experiments (c, d, f and g). Horizontal bars represent the mean of the points shown (c, d, e and j).
We started treatment with IL-22.Fc or vehicle control (PBS) intraperitoneally one day before C. rodentium infection and maintained treatment twice a week on days 2, 6, 9 and 13 p.i. (Fig. 2b). Exogenous IL-22.Fc reduced bacterial burden in the feces (Fig. 2c) on day 5 p.i. and reduced the endoscopic MEDAI compared with vehicle control on day 7 p.i. (Fig. 2d and f). Endoscopic measures of exudate, granularity and vascularity were all improved in IL-22.Fc-treated mice (Fig. 2d and f). Histologic evaluation confirmed a significant reduction in mucosal inflammation and hyperplasia in the distal and mid colon (Fig. 2e and f). IL-22 treatment also prevented the goblet cell loss, albeit with variability in the degree of amelioration (Supplementary Fig. 2c). IL-22.Fc treatment significantly enhanced survival in C. rodentium-infected FVB mice (Fig. 2g). Untreated and PBS treated mice lost significant more body weight than naïve animals on day 9 p.i., demonstrating the severity of disease in the infected animals (Supplementary Fig. 2d). Although the average body weights of IL-22 treated group were higher than those of untreated or vehicle control groups, the differences were not statistically significant consistent with other reports where IL-22 treatment had no impact on body weight34,35; however, this is in contrast to studies where exogenous IL-22 did improve body weight indicating that effect of IL-22 on body weight is context dependent36,37. Co-housing the IL-22 treated mice with vehicle treated mice conferred partial protection on vehicle treated mice (Supplementary Fig. 2e) consistent with the role of IL-22 in altering the endogenous microbiota to favor protection from pathogenic bacterial species15.
IL-22 drives host defense by eliciting the production of AMPs such as REG3B and REG3G in response to microbial infection4. Consistent with these published observations, we found that REG3B and REG3G protein levels were increased in IL-22.Fc-treated animals compared to vehicle control treated animals (Fig. 2h and j). Finally, to phenotypically assess the differentiation of intestinal epithelium, we evaluated expression of PHGR1 protein, which shows highest expression in the most differentiated cells of the human intestinal epithelium by IHC38. We found that PHGR1 was decreased in whole colonic tissue in infected untreated mice and IL-22.Fc-treatment restored the expression of PHGR1 to levels comparable to naïve mice (Fig. 2i and j). As such, IL-22 treatment potentiated host-defense, reduced disease severity and rescued FVB mice from C. rodentium infection induced lethality.
CITE-seq analysis revealed the emergence of undifferentiated intermediate IECs during disease, resolved the MoA of IL-22 in vivo and identified parallels between C. rodentium-induced colitis and IBD
Overt epithelial hyperplasia in FVB mice during C. rodentium infection likely interferes with normal epithelial differentiation, resulting in undifferentiated IECs akin to those reported in human samples5. Moreover, how IL-22 treatment regulates the function of various IEC lineages in vivo is not well understood. Thus, using single-cell CITE-seq technology, we determined whether there was heterogeneity in IECs in various treatment groups, and determined how IL-22 treatment altered their transcriptomic signature.
Colon cells were isolated on day 12 p.i. and processed for CITE-seq. We identified and visualized 24 cell clusters by Uniform Manifold Approximation and Projection (UMAP) (Fig. 3a). We first performed automatic cluster annotation using SingleR39 by comparing transcriptomic similarities of single cells from individual clusters to reference databases. Based on the SingleR analysis (shown in Supplementary Table 2), we identified 8 different cell types distributed across various clusters: 1) epithelial cells, 2) macrophages/monocytes, 3) dendritic cells, 4) T/NKT/Innate Lymphoid Cells, 5) B cells, 6) fibroblasts, 7) endothelial cells, and 8) stromal cells. For further analyses we focused on epithelial cell clusters (Clusters 0, 12, 16, 22 and 23), which expressed higher levels of EPCAM protein detected using a CITE-seq specific barcoded antibody (Fig. 3a, inset). Based on similarities in the transcriptomic signature, we annotated mouse IEC clusters by transferring cell type labels from a UC study to mouse IEC Clusters 0, 12, 16 and 22, but not Cluster 23 due to the smaller number of cells compared to other clusters, which precluded statistical analysis (Fig. 3a; Supplementary Table 4)5. We found that the majority of cells in Clusters 0, 12, 16 and 22 were undifferentiated cells, goblet cells, colonocytes and enteroendocrine cells (EECs), respectively (Fig. 3a; Supplementary Table 4). All the clusters shared similar cell type markers as their counterparts in human tissues5 (Supplementary Fig. 3a), thus confirming our cell type label transfer.

CITE-seq was conducted on lamina propria cells isolated from various treatment groups – infected (untreated, vehicle or IL-22.Fc treated) or naïve mice (n = 5 mice per group). a Integrated UMAP clustering of all different treatments highlighting the epithelial cell clusters that can be distinguished in the feature plot by expression of EPCAM antibody derived tag (inset). Clusters 0, 12, 16, 22 and 23: epithelial cells; Clusters 1, 3 and 9: macrophages/monocytes; Clusters 2, 8, 11 and 19: dendritic cells; Cluster 4: T cells, NKT cells and innate lymphoid cells; Clusters 5, 6 and 14: B cells; Clusters 7 and 10: fibroblasts; Clusters 15 and 21: endothelial cells; Cluster 17: stromal cells; Clusters 13, 18 and 20 contained mixed populations. b Pseudotime trajectory analysis of the IECs with stem cells (white), intermediate cells (Branched [B] or transitional [T]; grey), colonocytes (green), and goblet cells/EECs (blue). c Distribution of IEC types identified using pseudotime trajectory analysis in the various treatment groups.
Next, we performed pseudotime trajectory analyses to understand changes in IEC differentiation states in various treatment groups. In this analysis, we were unable to distinguish between goblet cells and EECs in Cluster 22 because of its small size, heterogeneous composition and due to overlapping secretory mechanisms between goblet cells and EECs, consistent with observations in human studies5. In all groups, trajectory analyses revealed a dichotomous trajectory of IECs developing from stem cells at Milestone S into two distinct differentiation lineages, i.e., absorptive, and secretory cells ending at Milestones C and G, respectively (Fig. 3b), as reported previously5. To elucidate the differentiation state of various clusters, we investigated their distribution within the trajectory. The majority of Cluster 16 was present at colonocyte Milestone C whereas Cluster 12 and Cluster 22 were at goblet cells/EECs Milestone G (Fig. 3b, c; Supplementary Table 5). Notably, after C. rodentium infection, intermediate states (Branching [B]/Transitional [T]) were present in both the colonocyte as well as goblet/EEC clusters in untreated or vehicle treated mice, indicative of infection-induced incomplete differentiation from progenitor cells (Fig. 3c; Supplementary Table 5). IL-22.Fc treatment restored the distribution of cells in the colonocyte cluster to a pre-disease state suggesting that Cluster 16 was highly responsive to IL-22. Using Cluster 12/22 and 16 as references, we studied the distribution of cell types within Cluster 0 and found that it contained mostly undifferentiated cells (i.e., stem cells or intermediate cells), some colonocytes but not goblet cells/EECs (Fig. 3b, c; Supplementary Table 5). Finally, to appreciate the degree of alteration in the overall landscape of IEC differentiation during each treatment condition, we combined the data from IEC clusters. After infection, there was an increase in the percentage of intermediate cells from 37.0% in naïve FVB mice to 51.2% in C. rodentium-infected FVB mice (Fig. 3c). IL-22.Fc treatment decreased the prevalence of intermediate cells to 28.3% compared with vehicle controls at 47.0% (Fig. 3c). To further understand the nature of intermediate IECs, we isolated Cluster 0 cells from infected untreated FVB mice in silico, since majority of the intermediate cells were undifferentiated Cluster 0 cells (Fig. 3c; Supplementary Table 5). We identified four subclusters in total (Supplementary Fig. 3b). The UC study reports five sub-cell types from human undifferentiated IECs including stem cells, early transit-amplifying cells, cell-cycle cluster cells (transit-amplifying-like-cells with high expression of cell cycle genes), absorptive progenitor cells and secretory progenitor cells5. We, therefore, annotated subclusters of the mouse undifferentiated cells by transferring sub-cell type labels from the UC study to mouse cells. Interestingly, we found all the four mouse subclusters were mixed cell populations of early transit-amplifying cells and absorptive progenitor cells (Supplementary Table 6).
In summary, we identified mouse IEC clusters that were akin to those observed in human samples5, thus underscoring the translatability of this model to human disease. Infection increased the frequency of intermediate IECs that have not completed their differentiation into terminal lineages critical for intestinal epithelial function, which likely contributed to disease development. IL-22.Fc treatment prevented the accumulation of intermediate IECs that occurred during disease.
CITE-seq analysis of C. rodentium-induced colitis following IL-22 treatment revealed disease-relevant changes in gene expression of specific epithelial cell populations that were similar to pathogenic mechanisms in IBD patients
Next, we asked whether specific IEC clusters responded differently to IL-22.Fc treatment versus vehicle in diseased mice. Cluster 22 emerged in C. rodentium-infected FVB mice, albeit with very low numbers (Fig. 3c), and therefore, we focused on IEC Clusters 0, 12, and 16 for analyses of differentially expressed genes (DEGs) comparing IL-22 treated mice to vehicle (Fig. 4a–d) and untreated infected mice to naïve (Supplementary Fig. 4a–d). When compared to naïve mice, a heat map of the overlapping DEGs revealed that most, but not all, of the disease associated changes were reversed by IL-22.Fc treatment (Supplementary Fig. 3c). Among the top 50 DEGs, we identified several markers of terminally differentiated IECs as well as solute transporter genes (Supplementary Table 7). Some solute transporter changes overlap with those found in whole tissue microarray studies reported previously10, thus validating our transcriptomics analyses. Next, we assessed the differential expression of genes involved in intestinal homeostasis, some of which were highlighted in Fig. 1c.

Heat maps of differentially expressed genes in IL-22.Fc treated mice compared to vehicle in various IEC clusters: IEC differentiation markers (a), AMPs (b), mucous layer genes (c), solute transporters (d). Clusters 0, 12 and 16 were undifferentiated cells, goblet cells and colonocytes, respectively.
Analyses of the differentiation markers reported in human IECs5 revealed that several genes had low to undetectable expression in mouse cells (Supplementary Fig. 3a). Among the detectable genes, we found that in infected untreated mice there was a decrease in markers that were highly expressed on differentiated human IECs, including Slc26a3, Car1, and Phgr1 in Clusters 0 and 16 when compared to naïve mice (Supplementary Fig. 4a). Of note, Phgr1 was among the top down-regulated genes in the undifferentiated and colonocyte cluster in diseased mice (Supplementary Table 7) validating the protein changes shown in Fig. 2i and j. IL-22.Fc treatment increased the expression of the majority of these differentiation markers including Slc26a3, Car1, Car4 and Phgr1 compared to vehicle (Fig. 4a). We did not observe a significant change in Alpi (Alkaline phosphatase, intestinal) either following the infection or after IL-22 treatment.
Among the AMPs, IL-22.Fc increased expression of Reg3b and Reg3g mRNA in all three IEC clusters (Fig. 4b), whereas Defb4540 and Rnase441 were only upregulated in undifferentiated cells. Strikingly, Wfdc2, a novel AMP reduced in UC patients5, was upregulated by IL-22.Fc in undifferentiated cells but was down-regulated in Cluster 16 colonocytes (Fig. 4b), thus highlighting cell type specific regulation of function. Since Cluster 0 is the largest IEC cluster (Supplementary Table 2), we speculate that there should be an increase in Wfdc2 expression in IL-22.Fc-treated animals.
For goblet cell differentiation and mucus layer integrity, we reviewed the genes involved in differentiation (Nupr1, Agr2, Klf4 and Elf3) or formation of critical components of the colonic mucus layer (Muc2, Muc3, Klk1, Oit1, Ceacam1, Mep1a, Lgals3, Dmbt1, Pigr and Pchd17)28,42,43,44,45,46,47,48. Except for Muc2, all the other genes were significantly up-regulated with IL-22.Fc treatment in undifferentiated stem cells (Fig. 4c). IL-22.Fc treatment significantly increased Muc2, Nupr1 and Agr2 in goblet cells suggesting that IL-22 drives differentiation and function of goblet cells. In contrast, Agr2 expression was decreased in colonocytes, consistent with its role in regulating goblet cell function44,46,47. Moreover, differential contributions of various epithelial cell clusters in establishing the mucus layer were evident by Klk1 and Oit1 induction in goblet cells and Ceacam1 induction in colonocytes (Fig. 4c).
Within the solute transporters (reviewed in27) we observed a decrease in several solute transporters in undifferentiated cells and colonocytes (Supplementary Fig. 4d), including Slc26a2 which was reduced in UC patient colonocytes compared to controls5. Compared to vehicle, IL-22.Fc treatment significantly: 1.) up-regulated several chloride absorptive transporters (Slc26a3, Slc26a2, Car1, Car2 and Car4), 2.) decreased chloride secretory transporters (Slc12a2) and 3.) decreased several Na+/K+-ATPase components and regulators (Atp1b1, Atp1b2, Fxyd3, Fxyd4 and Fxyd5) in Cluster 0 undifferentiated cells (Fig. 4d). Notably, IL-22 treatment resulted in distinct changes in goblet cells and colonocytes. Car1 and Car4 were significantly upregulated in colonocytes but not goblet cells, whereas Slc12a2 was significantly downregulated in goblet cells but not colonocytes. Finally, Slc26a2 was increased in undifferentiated cells and goblet cells after treatment (Fig. 4d), suggesting that IL-22 treatment should improve Cl− absorption49,50.
In summary, we furthered the understanding of IL-22 MoA at the single-cell level by elucidating its differential effect on undifferentiated IECs, goblet cells and colonocytes.
Exogenous IL-22 prevented development of impaired intestinal ion transport in C. rodentium-infected FVB mice
Previous studies reported a fatal diarrhea associated with impaired intestinal ion transport in FVB mice during C. rodentium infection leading to reduced serum levels of Na+ and Cl− ions indicative of hyponatremia and hypochloremia, respectively9. Apical SLC26A3 couples with CAR proteins to mediate absorption of Cl− from the intestinal lumen51,52. Solute transporters such as SLC26A3 play critical roles in maintaining Cl− balance in humans and mice53,54. While we observed the transcriptional regulation of several solute transporters by IL-22.Fc treatment at the resolution of single IECs (Fig. 4d), in order to determine if it alters the protein levels of these solute transporters in the whole tissue and levels of Na+ and Cl−, we conducted proteomics and western blots on colonic tissue and measured serum and fecal levels of Na+ and Cl− from IL-22.Fc- versus vehicle-treated mice. We assessed these changes between day 8 and day 11 p.i., time-points when we observed maximal disease activity.
Proteomics revealed that SLC26A3 and CAR4 were among the top up-regulated proteins by IL-22 treatment, when compared to vehicle-treated mice (Fig. 5a). Additionally, IL-22 treatment led to significant induction of serum amyloid A 2 (SAA2) and REG3β (Fig. 5a) consistent with previous studies in mouse models and clinical studies17,19. IL-22 treatment also increased protein expression of several mucus layer components28, including IgA heavy chain (IGHA), EEC-derived hormone Chromogranin A (CHGA), and extracellular matrix proteins Decorin (DCN) and Lumican (LUM) (Supplementary Fig. 4e), thus providing additional information on the MoA of IL-22 in reinforcing epithelial barrier function. A heat map highlighting changes of differentially expressed proteins showed that even as early as day 8 p.i., in some of the IL-22.Fc-treated mice, the protein expression changes were similar to that seen in naïve mice (Supplementary Fig. 2a). 3 out of 10 mice that were comparable to naïve mice also had the lowest amount of disease activity by histology for both the inflammation as well as hyperplasia scores (shown in Fig. 2e), thus correlating clinical disease read-outs with proteomics expression.

a Volcano plot depicting protein expression changes on day 8 p.i. in IL-22.Fc treated mice compared to vehicle controls using MS based proteomics. b, c Immunoblot analyses of CAR1, CAR2, CAR4, SLC26A3, SLC12A2, ATP1B2, ATP1B1 and GAPDH (loading control) in various treatment groups. Data from individual mice shown in (b) and densitometric quantification shown in (c). d Sodium and chloride levels in fecal samples or in the serum on day 11 p.i. in various treatment groups (n = 10 mice per group). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 determined as follows: one-way ANOVA with Dunnett’s multiple comparisons test (c, d). Horizontal bars represent the mean of the points shown (c, d).
We confirmed proteomics changes in solute transporters using western blots at the peak of disease on day 13 p.i. In vehicle-treated C. rodentium-infected FVB mice, we observed statistically significant down-regulation of CAR 1/2/4 and SLC26A3 as well as up-regulation of SLC12A2 (Fig. 5b, c). Furthermore, ATP1B2 was increased while ATP1B1 was not altered after C. rodentium infection (Fig. 5b, c). Together, these data suggested that it was likely that C. rodentium infection would be associated with a reduced ability to retain Cl−. All these diseases associated changes were prevented by IL-22.Fc administration (Fig. 5b, c). Moreover, protein levels of these transporters were restored to levels in naïve mice, except CAR4, where we observed 3-fold higher levels compared to naive mice (Fig. 5b, c).
Next, we measured Na+ and Cl− levels in circulation and feces. Compared to naïve mice, we observed statistically significant increases in Na+ and Cl− in the feces and reduced levels in the serum after C. rodentium infection (Fig. 5d). Moreover, at the same time points, untreated FVB mice had dehydration symptoms with increased levels of serum total protein, including albumin (Fig. 5d). IL-22.Fc treatment of C. rodentium-infected FVB mice prevented hyponatremia and hypochloremia as well as dehydration when compared to vehicle-treated animals (Fig. 5d). In C. rodentium-infected mice treated with IL-22.Fc, Na+ and total protein levels in the serum were comparable to naïve mice (Fig. 5d). In sum, the functional consequence of the changes in solute transporters in C. rodentium-infected FVB mice treated with IL-22.Fc was reflected in the restoration of homeostatic levels of Na+ and Cl− in the circulation to levels observed in naïve mice.
Discussion
Molecular changes in intestinal immune cells, stromal cells and epithelial cells in IBD has provided valuable insight into mechanisms that contribute to dysregulated intestinal homeostasis and barrier immunity5,55,56. Defects in epithelial cell function has been a challenge to study in pre-clinical models of colitis and bridging this gap is essential to understand the MoA of mucosal healing therapies for IBD such as IL-22. The multifaceted biology of IL-22 positions it as a central regulator of intestinal tissue homeostasis and necessitates an understanding of its impact in vivo on various IEC subsets. In this study, we demonstrated parallels between epithelial dysfunction reported in human IBD and C. rodentium-induced infectious colitis in FVB mice, albeit the former is a chronic disease while the latter is an acute model. Moreover, IL-22 treatment prevented the epithelial dysfunction reported in mice susceptible to C. rodentium infection and rescued mice from lethal diarrhea by preventing dysregulation of the solute transporter genes that are involved in Na+/Cl− transport in the intestinal epithelium. Interrogation of C. rodentium induced colitis using CITE-seq and proteomics offered us valuable insight into the translatability of the C. rodentium model to human disease and allowed us to understand the different IEC types and their response to IL-22 treatment in the model.
Striking similarities were observed between human IBD and the C. rodentium infectious colitis model. In C. rodentium infected mice, disease severity ranged from extensive mucosal hyperplasia and goblet cell loss in the distal colon, to milder disease in the mid colon characterized by heterogeneous loss of goblet cells, such that some crypts had more severe loss compared to other crypts, akin to what has been observed in IBD patients57. Comparison of whole tissue proteomics profiling at the peak of disease in the C. rodentium mouse model, combined with our previously published proteomics data in UC and CD tissues20, revealed a similar pattern of protein dysregulation. Compared to controls, in diseased human IBD and mouse colitis tissues, we observed down-regulation of proteins required for epithelial function and barrier immunity (such as SLC26A3, CAR4 and MUC2) as well as reduction in phenotypic markers of intestinal epithelial differentiation (such as CAR1, CAR4, SLC26A3, PHGR1 and SELENBP1)5. With CITE-seq, we identified IEC subsets in mice that share common key genes with their counterparts in human tissues. Moreover, we found that diseased mice had an expansion of intermediate IECs with a gene signature that overlaps with ‘undifferentiated’ IECs in UC patients5. Further analyses showed that the composition of the mouse intermediate cells overlapped with early transit-amplifying cells and absorptive progenitor cells of human undifferentiated IECs. Differentiation of colonocytes requires loss of Wnt signals58,59. Since FVB mice are reported to have excessive activation of Wnt pathway following C. rodentium infection11, the expansion of transit-amplifying cells and absorptive progenitor cells in the infected FVB might result in defective terminal differentiation of colonocyte due to unrestrained Wnt signaling. Therefore, the absence of intermediate cells in IL-22 treated mice is likely a consequence of the inhibitory effects of IL-22 on Wnt signaling12 that drives the differentiation of intermediate cells into colonocytes. In addition to IECs, we also observed shared cellular properties and functions between T cells in C. rodentium model and human CD patients. Thus, the underlying mechanisms in response to microbial dysbiosis in mice and humans share some overlapping mechanisms that regulate epithelial and T cell function; as such, the C. rodentium colitis in FVB mice models the role of microbial dysbiosis in IBD.
We showed that reduced levels of sodium and chloride ions corresponded with disease severity in the C. rodentium model of colitis. It is tempting to speculate that this process could be linked to the extent of inflammation. In vitro systems using human IECs previously demonstrated the reciprocal regulation of SLC26A3 by pro-inflammatory cytokine TNFα, and vice versa60. Whether this occurs on primary IECs and in vivo has not yet been determined. Due to the clinical success of TNFα neutralizing therapies in IBD, understanding the contribution of TNFα in regulating physiological IEC biology will further our understanding of the pathophysiology of IBD. Exogenous IL-22 prevented hypochloremia and hyponatremia by preventing disease-associated changes in transporters that maintain physiological solute balance, notably SLC26A3 and CAR4. IL-22.Fc treatment restored the total protein levels of SLC26A3 back to levels observed in naïve mice and IL-22.Fc increased CAR4 protein expression to levels greater than naïve mice, suggesting that CAR4 might be directly regulated by IL-22. Indeed, a STAT3 binding site is predicted in Car4 by the TRANSFAC database61, making Car4 a potential new target of IL-22 signaling. Slc26a3 deficiency in mice or loss of function mutations in humans results in congenital chloride diarrhea, underscoring its relevance in regulating physiological levels of Cl−53,54,62. While Car4 deficiency does not present with an overt homeostatic phenotype in gastrointestinal (GI) tissues, its down-regulation increases the risk of colorectal cancer due to an increase in Wnt signaling and gain of function studies reverses the phenotype63. Down-regulation of Car4 is observed in the TCRα deficient spontaneous colitis model but is not observed in the TCT or DSS colitis models64. Interestingly, local gene delivery of Il22 ameliorates the colitis of TCRα deficient mice36. Future studies are needed to explore the direct role of IL-22 in modulating expression of solute transporters, in particular CAR4.
Profound epithelial protective effects of IL-22 treatment were observed after infection with C. rodentium in susceptible FVB mice that have defects in epithelial function. We did not observe the protective effect of IL-22 in infected C57BL/6 mice (Supplementary Fig. 5). One key difference is that C57BL/6 mice lack the Pla2g2a gene and do not overexpress Rspo2 in response to C. rodentium infection, compared to FVB mice11,25. Both PLA2G2A and RSPO2 increase Wnt signaling and proliferation. A Wnt signaling inhibitor ameliorates C. rodentium induced disease in the other susceptible C3H strain11. Thus, we conclude that underlying host genetics influenced the outcome of IL-22 treatment.
Our studies indicated that the pro-differentiation MoA of IL-22 was apparent in a model with defects in barrier function. These data indicated that the mode of action for IL-22 was context-dependent. IL-22 provides anti-microbial immunity in response to several infection models, but not all4. Most of these studies relied on genetic deficiencies or antibody neutralization of IL-22, which do not necessarily predict the behavior of agonistic IL-22.Fc treatment. However, treatment with IL-22 is protective in a DSS-induced epithelial damage model in C57BL/6 mice17. In IBD tissues, we and others have observed areas of erosion interspersed with areas of hyperplasia (shown here57). Based on this, we speculate that, depending on the underlying genetics and local cues, IL-22 treatment might heal the barrier disruption of IBD by inducing proliferation and promoting survival of IECs but could also drive differentiation within the hyperproliferative areas.
We elucidated the IL-22-mediated transcriptional changes in distinct IEC subsets. Several lines of evidence confirmed the induction of known targets of IL-22 and identified new molecules that reinforce barrier immunity. Consistent with previous studies of homeostatic, chemical injury-induced and spontaneous colitis mouse models treated with IL-22.Fc or colonic Il22 gene delivery17,36,65,66, we observed induction of REG3β, REG3γ, MUC2 and SAA2 in colonic tissues with IL-22.Fc treatment. Reg3b and Reg3g were induced in all IEC clusters. In undifferentiated IECs, IL-22.Fc treatment increased several other AMPs, including Wfdc2 that regulates homeostatic barrier integrity5, as well as numerous genes that reinforce the protective effects of the mucus layer, including Dmbt1. DMBT1, aka Muclin, is a GI restricted protein induced by NOD2 signaling and prevents invasion of IECs by pathogenic bacteria67. A deletion variant of human DMBT1 impairs its activity and is associated with CD68,69. Importantly, IL-22 has been shown to induce DMBT1 via STAT3 signaling in a SW403 human colon epithelial cell line70, thereby adding to the ability of IL-22 to prevent microbial dysbiosis71.
To the best of our knowledge, this is the first demonstration of a pre-clinical model that can be used to study the dysregulation of intestinal epithelial cell function. Single-cell IEC transcriptomics in the C. rodentium colitis in FVB mice aligned with published human single cell IEC data5 as well as with our previous proteomics analyses in IBD tissues20. Moreover, we extended the role of IL-22 in regulating gut homeostasis beyond its role in inducing anti-microbial peptide and mucin production to include critical solute transporters. Finally, we observed the concordance between transcriptomics and proteomics for several genes/proteins critical for intestinal homeostasis, thus underscoring the importance of multi-omics approaches to further our understanding of the underlying disease mechanisms in mouse models as well as human disease.
Methods
Methods are in Supplemental Materials.
References
de Souza, H. S. & Fiocchi, C. Immunopathogenesis of IBD: current state of the art. Nat. Rev. Gastroenterol. Hepatol. 13, 13–27 (2016).
Caruso, R., Lo, B. C. & Nunez, G. Host-microbiota interactions in inflammatory bowel disease. Nat. Rev. Immunol. 20, 411–426 (2020).
Uhlig, H. H. & Powrie, F. Translating Immunology into Therapeutic Concepts for Inflammatory Bowel Disease. Annu Rev. Immunol. 36, 755–781 (2018).
Keir ME, Yi TS, Lu TT, Ghilardi N. The role of IL-22 in intestinal health and disease. J. Exp. Med. 217, e20192195 (2020).
Parikh, K. et al. Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature 567, 49–55 (2019).
Kiesler, P., Fuss, I. J. & Strober, W. Experimental Models of Inflammatory Bowel Diseases. Cell Mol. Gastroenterol. Hepatol. 1, 154–170 (2015).
Collins, J. W. et al. Citrobacter rodentium: infection, inflammation and the microbiota. Nat. Rev. Microbiol 12, 612–623 (2014).
Gevers, D. et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe .15, 382–392 (2014).
Borenshtein, D. et al. Decreased expression of colonic Slc26a3 and carbonic anhydrase iv as a cause of fatal infectious diarrhea in mice. Infect. Immun. 77, 3639–3650 (2009).
Borenshtein, D. et al. Diarrhea as a cause of mortality in a mouse model of infectious colitis. Genome Biol. 9, R122 (2008).
Papapietro, O. et al. R-spondin 2 signalling mediates susceptibility to fatal infectious diarrhoea. Nat. Commun. 4, 1898 (2013).
Zha, J. M. et al. Interleukin 22 Expands Transit-Amplifying Cells While Depleting Lgr5(+) Stem Cells via Inhibition of Wnt and Notch Signaling. Cell Mol. Gastroenterol. Hepatol. 7, 255–274 (2019).
Lindemans, C. A. et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 528, 560–564 (2015).
Zenewicz, L. A. et al. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 29, 947–957 (2008).
Tsai, P. Y. et al. IL-22 Upregulates Epithelial Claudin-2 to Drive Diarrhea and Enteric Pathogen Clearance. Cell Host Microbe. 21, 671–681 (2017). e674.
Pham, T. A. et al. Epithelial IL-22RA1-mediated fucosylation promotes intestinal colonization resistance to an opportunistic pathogen. Cell Host Microbe. 16, 504–516 (2014).
Stefanich, E. G. et al. Pre-clinical and translational pharmacology of a human interleukin-22 IgG fusion protein for potential treatment of infectious or inflammatory diseases. Biochem Pharm. 152, 224–235 (2018).
Ouyang, W. & O’Garra, A. IL-10 Family Cytokines IL-10 and IL-22: from Basic Science to Clinical Translation. Immunity 50, 871–891 (2019).
Rothenberg, M. E. et al. Randomized Phase I Healthy Volunteer Study of UTTR1147A (IL-22Fc): A Potential Therapy for Epithelial Injury. Clin. Pharm. Ther. 105, 177–189 (2019).
Jin, L. et al. Integrative Analysis of Transcriptomic and Proteomic Profiling in Inflammatory Bowel Disease Colon Biopsies. Inflamm. Bowel Dis. 25, 1906–1918 (2019).
Borenshtein, D., Nambiar, P. R., Groff, E. B., Fox, J. G. & Schauer, D. B. Development of fatal colitis in FVB mice infected with Citrobacter rodentium. Infect. Immun. 75, 3271–3281 (2007).
Omidbakhsh, A., Saeedi, M., Khoshnia, M., Marjani, A. & Hakimi, S. Micro-RNAs -106a and -362-3p in Peripheral Blood of Inflammatory Bowel Disease Patients. Open Biochem J. 12, 78–86 (2018).
Gersemann, M. et al. Differences in goblet cell differentiation between Crohn’s disease and ulcerative colitis. Differentiation 77, 84–94 (2009).
Strugala, V., Dettmar, P. W. & Pearson, J. P. Thickness and continuity of the adherent colonic mucus barrier in active and quiescent ulcerative colitis and Crohn’s disease. Int J. Clin. Pr. 62, 762–769 (2008).
Schewe, M. et al. Secreted Phospholipases A2 Are Intestinal Stem Cell Niche Factors with Distinct Roles in Homeostasis, Inflammation, and Cancer. Cell Stem Cell. 19, 38–51 (2016).
Lyons J, et al. Integrated in vivo multiomics analysis identifies p21-activated kinase signaling as a driver of colitis. Sci. Signal. 11, eaan3580 (2018).
Thiagarajah, J. R. & Verkman, A. S. Chloride channel-targeted therapy for secretory diarrheas. Curr. Opin. Pharm. 13, 888–894 (2013).
van der Post, S. et al. Structural weakening of the colonic mucus barrier is an early event in ulcerative colitis pathogenesis. Gut 68, 2142–2151 (2019).
Odenwald, M. A. & Turner, J. R. The intestinal epithelial barrier: a therapeutic target? Nat. Rev. Gastroenterol. Hepatol. 14, 9–21 (2017).
Zihni, C., Mills, C., Matter, K. & Balda, M. S. Tight junctions: from simple barriers to multifunctional molecular gates. Nat. Rev. Mol. Cell Biol. 17, 564–580 (2016).
Peake, M. A. et al. Identification of a transcriptional signature for the wound healing continuum. Wound Repair Regen. 22, 399–405 (2014).
Martin, J. C. et al. Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy. Cell 178, 1493–1508 (2019).
Zheng, Y. et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 14, 282–289 (2008).
Ota, N. et al. IL-22 bridges the lymphotoxin pathway with the maintenance of colonic lymphoid structures during infection with Citrobacter rodentium. Nat. Immunol. 12, 941–948 (2011).
Kim S, et al. Amelioration of DSS-induced Acute Colitis in Mice by Recombinant Monomeric Human Interleukin-22. bioRxiv 22:e26 (2022).
Sugimoto, K. et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Invest 118, 534–544 (2008).
Cox, J. H. et al. Opposing consequences of IL-23 signaling mediated by innate and adaptive cells in chemically induced colitis in mice. Mucosal Immunol. 5, 99–109 (2012).
Oltedal, S. et al. Expression profiling and intracellular localization studies of the novel Proline-, Histidine-, and Glycine-rich protein 1 suggest an essential role in gastro-intestinal epithelium and a potential clinical application in colorectal cancer diagnostics. BMC Gastroenterol. 18, 26 (2018).
Aran, D. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 20, 163–172 (2019).
Schutte, B. C. et al. Discovery of five conserved beta-defensin gene clusters using a computational search strategy (vol 99, pg 2129, 2002). P Natl Acad. Sci. 99, 14611–14611 (2002).
Murtha, M. J. et al. Insulin receptor signaling regulates renal collecting duct and intercalated cell antibacterial defenses. J. Clin. Invest. 128, 5634–5646 (2018).
Ng, A. Y. et al. Inactivation of the transcription factor Elf3 in mice results in dysmorphogenesis and altered differentiation of intestinal epithelium. Gastroenterology 122, 1455–1466 (2002).
Gregorieff, A. et al. The ets-domain transcription factor Spdef promotes maturation of goblet and paneth cells in the intestinal epithelium. Gastroenterology 137, 1333–1345 (2009).
Park, S. W. et al. The protein disulfide isomerase AGR2 is essential for production of intestinal mucus. Proc. Natl Acad. Sci.106, 6950–6955 (2009).
Ghaleb, A. M., McConnell, B. B., Kaestner, K. H. & Yang, V. W. Altered intestinal epithelial homeostasis in mice with intestine-specific deletion of the Kruppel-like factor 4 gene. Dev. Biol. 349, 310–320 (2011).
Bergstrom, J. H. et al. AGR2, an endoplasmic reticulum protein, is secreted into the gastrointestinal mucus. PLoS One.9, e104186 (2014).
McCauley, H. A. & Guasch, G. Three cheers for the goblet cell: maintaining homeostasis in mucosal epithelia. Trends Mol. Med. 21, 492–503 (2015).
Johansson, M. E. & Hansson, G. C. Immunological aspects of intestinal mucus and mucins. Nat. Rev. Immunol. 16, 639–649 (2016).
Haila, S. et al. SLC26A2 (diastrophic dysplasia sulfate transporter) is expressed in developing and mature cartilage but also in other tissues and cell types. J. Histochem Cytochem. 49, 973–982 (2001).
Alper, S. L. & Sharma, A. K. The SLC26 gene family of anion transporters and channels. Mol. Asp. Med. 34, 494–515 (2013).
Sterling, D., Brown, N. J. D., Supuran, C. T. & Casey, J. R. The functional and physical relationship between the DRA bicarbonate transporter and carbonic anhydrase II. Am. J. Physiol.-Cell Ph. 283, C1522–C1529 (2002).
Kato, A. & Romero, M. F. Regulation of electroneutral NaCl absorption by the small intestine. Annu Rev. Physiol. 73, 261–281 (2011).
Schweinfest, C. W. et al. slc26a3 (dra)-deficient mice display chloride-losing diarrhea, enhanced colonic proliferation, and distinct up-regulation of ion transporters in the colon. J. Biol. Chem. 281, 37962–37971 (2006).
Wedenoja, S. et al. Update on SLC26A3 mutations in congenital chloride diarrhea. Hum. Mutat. 32, 715–722 (2011).
Kinchen, J. et al. Structural Remodeling of the Human Colonic Mesenchyme in Inflammatory Bowel Disease. Cell 175, 372–386 (2018).
Smillie, C. S. et al. Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 178, 714–730 (2019).
DeRoche, T. C., Xiao, S. Y. & Liu, X. Histological evaluation in ulcerative colitis. Gastroenterol. Rep. 2, 178–192 (2014).
Yin, X. et al. Niche-independent high-purity cultures of Lgr5+ intestinal stem cells and their progeny. Nat. Methods. 11, 106–112 (2014).
Beumer, J. & Clevers, H. Cell fate specification and differentiation in the adult mammalian intestine. Nat. Rev. Mol. Cell Biol. 22, 39–53 (2021).
Ding, X. et al. Tumor necrosis factor-alpha acts reciprocally with solute carrier family 26, member 3, (downregulated-in-adenoma) and reduces its expression, leading to intestinal inflammation. Int J. Mol. Med. 41, 1224–1232 (2018).
Rouillard AD, et al. The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database (Oxford)2016, baw100 (2016).
Anbazhagan, A. N., Priyamvada, S., Alrefai, W. A. & Dudeja, P. K. Pathophysiology of IBD associated diarrhea. Tissue Barriers. 6, e1463897 (2018).
Zhang, J. et al. Carbonic anhydrase IV inhibits colon cancer development by inhibiting the Wnt signalling pathway through targeting the WTAP-WT1-TBL1 axis. Gut 65, 1482–1493 (2016).
Mizoguchi, E. et al. Colonic epithelial functional phenotype varies with type and phase of experimental colitis. Gastroenterology 125, 148–161 (2003).
Shih, V. F. et al. Homeostatic IL-23 receptor signaling limits Th17 response through IL-22-mediated containment of commensal microbiota. Proc. Natl Acad. Sci. 111, 13942–13947 (2014).
Hainzl, E. et al. Intestinal Epithelial Cell Tyrosine Kinase 2 Transduces IL-22 Signals To Protect from Acute Colitis. J. Immunol. 195, 5011–5024 (2015).
Rosenstiel, P. et al. Regulation of DMBT1 via NOD2 and TLR4 in intestinal epithelial cells modulates bacterial recognition and invasion. J. Immunol. 178, 8203–8211 (2007).
Renner, M. et al. DMBT1 confers mucosal protection in vivo and a deletion variant is associated with Crohn’s disease. Gastroenterology 133, 1499–1509 (2007).
Bikker, F. J. et al. The scavenging capacity of DMBT1 is impaired by germline deletions. Immunogenetics 69, 401–407 (2017).
Fukui, H. et al. DMBT1 is a novel gene induced by IL-22 in ulcerative colitis. Inflamm. Bowel Dis. 17, 1177–1188 (2011).
Bergstrom, J. H. et al. Gram-positive bacteria are held at a distance in the colon mucus by the lectin-like protein ZG16. Proc. Natl Acad. Sci.113, 13833–13838 (2016).
Acknowledgements
We thank former AbbVie employee Kristoff Homan for generation of the IL-22.Fc construct used in these studies, John Maull of AbbVie for proteomics sample preparation, Eunice Alejandria of AbbVie for clinical chemistry analyses, AbbVie employees Sam Karsen, Heather Knight, and Yingli Yang for histology and immunohistochemistry, as well as Sarah Wilson of AbbVie for helpful discussions.
Author information
Authors and Affiliations
Contributions
Conceptualization: Q.Z., R.G. Data collection/analysis: Q.Z., D.K., A.S.F, S.G., L.J., S.P., R.D., M.P., A.C., C.H., L.P., S.W., Y.B., J.P.C., R.G. Supervision: R.G., L.P., J.P.C., M.C.L., X.C., Y.T., A.J.S.S. Writing - original draft preparation: R.G., Q.Z., M.L., D.K., A.S.F, S.G., L.J., Y.B., J.P.C. Writing - review and editing: R.G., Q.Z., M.C.L., L.P., S.W., B.L.M., X.C., R.K.
Corresponding author
Ethics declarations
Competing interests
Q.Z., D.K., A.S.F., L.J., S.P., M.P., A.C., C.H., Y.T., R.K., L.P., A.J.S., S.W. and Y.B. are employees of AbbVie. S.G., R.D., B.L.M., X.C., M.C.L., J.P.C. and R.G. were employees of AbbVie at the time of the study. The design, study conduct, and financial support for this research were provided by AbbVie. AbbVie participated in the interpretation of data, review, and approval of the publication.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhu, Q., Korenfeld, D., Suarez-Fueyo, A. et al. Epithelial dysfunction is prevented by IL-22 treatment in a Citrobacter rodentium-induced colitis model that shares similarities with inflammatory bowel disease. Mucosal Immunol 15, 1338–1349 (2022). https://doi.org/10.1038/s41385-022-00577-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-022-00577-w
This article is cited by
-
Strategies for targeting cytokines in inflammatory bowel disease
Nature Reviews Immunology (2024)
