
Abstract
Self-replicating RNA viruses have been engineered as efficient expression vectors for vaccine development for infectious diseases and cancers. Moreover, self-replicating RNA viral vectors, particularly oncolytic viruses, have been applied for cancer therapy and immunotherapy. Among negative strand RNA viruses, measles viruses and rhabdoviruses have been frequently applied for vaccine development against viruses such as Chikungunya virus, Lassa virus, Ebola virus, influenza virus, HIV, Zika virus, and coronaviruses. Immunization of rodents and primates has elicited strong neutralizing antibody responses and provided protection against lethal challenges with pathogenic viruses. Several clinical trials have been conducted. Ervebo, a vaccine based on a vesicular stomatitis virus (VSV) vector has been approved for immunization of humans against Ebola virus. Different types of cancers such as brain, breast, cervical, lung, leukemia/lymphoma, ovarian, prostate, pancreatic, and melanoma, have been the targets for cancer vaccine development, cancer gene therapy, and cancer immunotherapy. Administration of measles virus and VSV vectors have demonstrated immune responses, tumor regression, and tumor eradication in various animal models. A limited number of clinical trials have shown well-tolerated treatment, good safety profiles, and dose-dependent activity in cancer patients.
Similar content being viewed by others
Introduction
Viral vectors have proven efficient in development of vaccines and therapeutics against infectious diseases and cancers [1]. A variety of viral vectors have been utilized including adenovirus, adeno-associated virus (AAV), herpes simplex virus (HSV), retroviruses, and lentiviruses. Only more recently, although being around for some time, self-replicating RNA viruses have received more attention. Self-replicating RNA viruses have some unique features, which make them attractive delivery vehicles for gene therapy and vaccine development [2]. The vectors possess a fairly good packaging capacity ranging from 4 to 6 kb of foreign genetic material. Self-replicating RNA viruses have a broad host range, which allows efficient delivery of RNA to mammalian cell lines, primary cells, and animal models. The RNA is directly released in the cytoplasm, where it is subjected to extensive amplification, giving these viruses the name self-replicating viruses (Fig. 1). The expression from a viral subgenomic promoter generates large amounts of recombinant protein, which can act as antigen for vaccine development or in case of expression of toxic genes, anti-tumor genes, or immunostimulatory genes can provide therapeutic effect against cancers. Due to the delivery in the form of RNA, which will degrade within 5–7 days, the expression is transient and the viral RNA or the gene of interest will not be integrated into the host cell genome. For these reasons, self-replicating RNA viruses are ideal for vaccine development and cancer therapy, which require a short boost of high-level transgene expression without being present for an extended time. However, in cases of chronic disease, which requires long-term, life-long transgene expression, AAV [3] and lentivirus [4] vectors are more suitable.

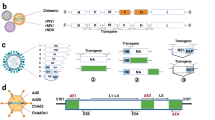
A Measles virus: the gene of interest is introduced either between the P and M genes or the H and L genes (maximum packaging capacity of 6 kb). B Vesicular stomatitis virus: for pseudotyped VSV the VSV-G gene is replaced by the foreign gene. Alternatively, foreign genes are introduced between the M and G genes and/or the G and L genes (maximum packaging capacity of 6 kb). C Rabies virus: the gene of interest is introduced between the N and P genes or between the G and L genes (maximum packaging capacity of 6 kb). CMV cytomegalovirus promoter; Fu fusion protein, G glycoprotein; Gol gene of interest; H hemagglutinin; L large protein; M matrix protein; N nucleocapsid; P phosphoprotein; T7 T7 RNA polymerase promoter; T7T T7 terminal sequence.
Self-replicating RNA viruses can be divided into two groups: Positive strand RNA viruses and negative strand RNA viruses. The polarity of the genome plays an important role. In the case of positive strand RNA viruses, protein translation from RNA can occur directly in the cytoplasm after viral delivery [5]. The RNA self-replication takes place from the replicase complex composed of the four non-structural proteins nsP1-4, which is estimated to generate 106 copies of subgenomic RNA per cell [6]. Expression systems based on Semliki Forest virus (SFV) [7], Sindbis virus (SIN) [8], and Venezuelan equine encephalitis virus (VEEV) [9] have demonstrated strong immune responses and protection against challenges in animal models [10]. However, this review focuses on negative strand RNA viruses. In contrast to the positive strand RNA viruses, the negative strand RNA viruses require the application of reverse genetics for their use as delivery vehicles [11]. In this review, the engineering of self-replicating negative strand RNA viral vectors is presented. Moreover, their applications for vaccine development against infectious diseases and cancers as well as their use in cancer therapy and cancer immunotherapy are discussed.
Engineering of expression vectors
Due to their negative polarity, measles virus (MV) and rhabdoviruses depend on reverse genetics for the engineering of recombinant expression systems [11]. Recovery of negative strand self-replicating RNA viruses from cDNA components or synthetic RNA as the initiation of replication requires de novo protein synthesis of its own RNA-dependent RNA polymerase (RdRp). In contrast to the replicase complex located in the non-structural genes of positive strand RNA viruses, the RdRp of negative strand RNA viruses is of structural origin and for instance, for VSV, the phosphoprotein (P) and large protein (L) constitute the RdRp [12].
A. Measles virus
In the case of MV, different rescue methods have been developed [13, 14]. HEK293 cells stably expressing the MV-N, MV-P, and T7 polymerase are co-transfected with a DNA plasmid carrying the full-length MV genome and the gene of interest and the T7 polymerase-driven DNA plasmid expressing MV-L [14, 15]. Alternatively, DNA plasmids expressing the MV-N, MV-P, and MV genome can be transfected into HEK293, which are superinfected with a replication-deficient vaccinia virus (MVA, modified vaccinia virus Ankara) expressing T7 polymerase. Furthermore, CMV-driven DNA plasmids expressing the MV genome and the RdRp components have been successfully used for MV rescue [16]. For recombinant protein expression from MV vectors, the gene of interest can be introduced either between the P and the matrix protein (M) of MV or alternatively between the hemagglutinin (H) and the L protein (Fig. 1A).
B. Rhabdoviruses
In the case of rhabdoviruses, expression systems have been engineered for VSV and rabies virus (RABV). For VSV both pseudotyped and recombinant VSV particles can be produced [17, 18]. Pseudotyped VSV can be generated in mammalian cells by transfection of DNA plasmids to replace the VSV glycoprotein (G) by a foreign envelope protein. Alternatively, foreign genes can be introduced between the M and G genes and/or the G and L genes (Fig. 1B). Transduction of mammalian cells with the pseudotyped VSV generates recombinant protein expression, but no virus progeny is produced [17]. In contrast, fully infectious VSV particles can be generated in mammalian producer cell lines, where the VSV-G envelope has been replaced by a foreign viral envelope [18]. Similar to MV vectors, rhabdovirus vectors have been engineered using reverse genetics-based on recombinant vaccinia virus vectors [19]. However, a vaccinia virus-free system has been engineered by the insertion of the VSV N, P, and L genes downstream of a T7 polymerase promoter and an internal ribosome entry site (IRES) [20]. The Maraba virus has also been engineered as a vaccine vector and due to its oncolytic activity, it has demonstrated potential in cancer treatment [21]. In the case of RABV, a vaccinia virus-free reverse genetics system has been engineered [22]. For recombinant protein expression, the genes of interest are introduced between the N and P genes or between the G and L genes in the RABV genome [23, 24] (Fig. 1C).
Negative strand self-replicating RNA virus vectors and infectious diseases
Negative strand self-replicating RNA viruses have been employed for vaccine development against infectious diseases. The general approach has been to engineer recombinant expression systems for overexpression of surface proteins of infectious agents as antigens with the aim to elicit immune responses and potential protection against challenges with lethal doses of pathogens. Below and in Table 1 are presented selected examples of negative strand RNA virus-based vaccine development against infectious diseases.
Due to several recent outbreaks, alphaviruses have been considered important targets for vaccine development [25, 26]. In this context, the envelope proteins of the Chikungunya virus (CHIKV) have been expressed from negative strand self-replicating RNA viral vectors. For instance, a chimeric VSV vector was applied for the expression of the CHIKV envelope polyprotein E3-E2-6K-E1. Immunization of mice elicited response of neutralizing antibodies [27]. Moreover, a single immunization with 1 × 107 VSV particles provided protection of mice challenged with lethal doses of CHIKV. In addition to VSV vectors, the attenuated MV Schwarz strain has been used for heterologous gene expression [28]. The whole subgenomic reading frame of the CHIKV structural genes (C-E3-E2-6K-E1) was introduced between the P and M proteins in the MV genome. Infection of Vero cells with the MV-CHIKV produced virus-like particles (VLPs), which could induce robust and protective humoral and cellular immune responses in immunized mice and furthermore protected mice from lethal challenges [29]. Moreover, macaques immunized with MV VLPs expressing the CHIKV capsid (C) and envelope proteins (E3-E2-6K-E1) generated strong immunogenicity and provided protection against lethal CHIKV challenges [30]. Furthermore, MV-CHIKV VLPs have been subjected to human clinical trials. In a randomized double-blind phase I clinical trial, a single dose provided a seroconversion rate of 44–92%, which reached 100% after a booster immunization [31]. The follow-up phase II trial elicited strong antibody responses and did not cause any serious adverse events [32]. Other alphaviruses such as Eastern equine encephalitis virus (EEEV) and Venezuelan equine encephalitis virus (VEEV) have been targeted for vaccine development using attenuated rhabdovirus vectors based on recombinant VSV and the serologically distinct Isfahan virus (rISFV) [33]. A new potential vaccine platform was established by rescuing rISFV from genomic DNA. A single immunization with rISFV expressing the EEEV and VEEV envelope proteins (E3-E2-6K-E1) elicited neutralizing antibody responses and protected mice from lethal challenges with EEEV and VEEV. Similar results were obtained for recombinant VSV-based EEEV and VEEV vaccines [33].
In the case of Lassa hemorrhagic fever (LHF), VSV vectors expressing the Lassa virus glycoprotein complex (GPC) showed protection of immunized guinea pigs against challenges with LASV strains from Liberia, Mali, and Nigeria [34]. Similarly, complete protection was observed against the Liberian LASV isolate in immunized macaques. Moreover, guinea pigs were vaccinated with a single dose of VSV-LASV GPC for the evaluation of induction of rapid and long-term immunity [35]. The study demonstrated that 100% protection against LSV challenges was obtained at 7- and 14-days post-immunization, whereas 83 and 87% protection was seen at 25-days and 6 months, respectively, after vaccination. Even one year after vaccination, the vaccination resulted in 71% protection in guinea pigs. In another study, the VSV-LASV GPC vaccine elicited both humoral and cellular immune responses and showed 100% protection in a non-human primate model [36]. In the case of RABV, the RABV G gene was replaced by the LASV-GPC gene [37]. Immunization of mice and guinea pigs with RABV-LASV-GPC elicited lasting humoral immune responses and provided protection against challenges with LASV. Attenuated MV has been utilized for the expression of LASV GPC or in combination with the LASV nucleoprotein (NP) [38]. Although MV-LASV-GPC elicited strong immune responses and protected cynomolgus monkeys from lethal challenges with LASV, the combination of LASV-GPC and NP showed superior efficacy. The recruitment for a randomized, placebo-controlled, dose-finding phase I clinical trial in healthy volunteers has been completed for the MV-LASV-GPC vaccine, but no data is available yet [39].
The dramatic Ebola virus (EBOV) epidemic with more than 11,000 fatalities in West Africa in 2013–2016 has surely accelerated the vaccine development. Two recombinant RABV vectors expressing the Zaire EBOV glycoprotein (ZGP) and the Sudan EBOV glycoprotein (SGP) genes, respectively, elicited neutralizing antibodies in immunized mice [40]. Moreover, dogs orally vaccinated with RABV-EBOV-ZGP developed long-lasting responses against ZEBOV. VSV vectors have been popular delivery vehicles for EBOV vaccines. VSV vectors expressing the EBOV glycoprotein (GP) were administered to cynomolgus macaques at a dose of 5 × 107 pfu [41]. Partial protection against EBOV was achieved when the immunization took place only 3 days before the challenge. On the other hand, immunization 7 days prior to challenge provided complete protection. Furthermore, immunization of macaques with 1 × 107 to 1 × 109 pfu of VSV-EBOV-GP resulted in complete protection against challenges with lethal doses of EBOV even after a single low dose of vaccine [42]. VSV vectors with the VSV glycoprotein (G) replaced by EBOV GP named VSV-ZEBOV were subjected to a single intraperitoneal administration of BALB/c with as few as 2 pfu, provided complete protection against challenges with lethal doses of EBOV [43]. In a similar way, a single intramuscular injection of VSV-ZEBOV elicited strong humoral and cellular immune responses in cynomolgus monkeys [44]. Moreover, immunization with 1000 pfu of VSV-ZEBOV protected monkeys from challenges with the Zaire EBOV strain. Clinical evaluation of the VSV-ZEBOV vaccine has been carried out. In a phase III clinical evaluation in 7651 individuals with suspected Ebola virus disease (EVD), 4123 persons were vaccinated immediately, and 3528 individuals received a delayed vaccination [45]. The efficacy of the vaccination was confirmed by no EVD cases detected in immediately vaccinated individuals and 16 EVD positive cases after delayed vaccination. The vaccine efficacy was confirmed in another phase III clinical study as no new EVD cases were detected in neither the 2119 immediately immunized nor the 2041 individuals, which were subjected to a 21-day delay in vaccination [46]. Both the FDA and the EMA have approved VSV-ZEBOV for EBOV vaccinations [47]. In the case of Marburg virus (MARV), the replacement of the VSV-G protein by the MARV GP provided complete protection against challenges with lethal doses of MARV in non-human primates [44].
Among flaviviruses, Dengue virus (DENV) has been a target for vaccine development applying MV vectors expressing tetravalent tandem repeats of envelope protein domain III (ED3) from DENV-1 to DENV-4 [48]. Immunization of hCD46 mice induced specific interferon-gamma (IFN-γ) and DENV- and MV-specific antibody responses. Moreover, the immunization provided protection against challenges with four DENV serotypes. VSV vectors have also been applied for DENV vaccine development. In this context, VSV expressing the DENV-2 pre-membrane (prM) and envelope (E) proteins was subcutaneously administered to BALB/c mice [49]. The immunization elicited anti-DENV-2 antibodies and protected mice against challenges with DENV-2. Vaccine development against another flavivirus, Zika virus (ZIKV), has also taken place. The chimeric VSV vector expressing the ZIKV prME proteins generated strong antibody responses and resulted in resistance against ZIKV infection in interferon receptor-deficient A129 mice [27]. Also inactivated RABV vectors have been engineered to express the ZIKV prME proteins [50]. BALB/c mice were immunized with RABV-ZIKV prME mixed with an ISA 201 VG and poly(I:C) adjuvant complex, which elicited both ZIKV and RABV titers greater than the protective titers. Therefore, it presents a promising approach for developing protection against both ZIKV and RABV infections. A live attenuated MV-based vector was engineered to express the ZIKV-E gene (MV-E2), which provided protection in immunized mice although complete viral clearance was not reached [51]. However, co-administration of the ZIKV non-structural 1 gene (NS1) from an MV vector showed complete ZIKV clearance. A dose-finding phase I trial was conducted with MV-ZIKA (MV-E2) in 48 volunteers, but no results are yet available [52]. Another phase I trial is in progress to evaluate the safety and immunogenicity of MV-ZIKA (MV-ZIKA-RSP) in 18–55-year-old volunteers [53].
In the context of vaccines developed against hepatotropic viruses such as hepatitis B virus (HBV) negative-strand self-replicating RNA viruses have been applied. For instance, expression of the HBV surface antigen (HBsAg) from an MV vector elicited strong immune responses and resulted in protection in 50% of immunized rhesus macaques [54]. Moreover, a VSV vector expressing the HBV middle envelope surface protein (MHBs) induced strong HBV-specific antibody responses in mice [55]. A single immunization induced CD8+ T-cell activation and protected mice against HBV challenge with hydrodynamic DNA transfection technique.
The development of HIV/AIDS vaccines has been an area of intensive research activity also including evaluation of negative-strand self-replicating RNA viruses. For instance, an MV vector expressing the HIV-Env gene generated neutralizing antibodies and cellular immune responses to both MV and HIV-Env in mice and macaques after a single immunization [56]. Moreover, the live attenuated MV strain has been utilized for the expression of HIV antigens. Recombinant MV-HIV Gag elicited humoral and cellular immune responses against MV and HIV and conferred protective immunity in vaccinated transgenic mice [57]. VSV vectors have also been used for HIV vaccine development [58]. Immunization of rhesus macaques with a combination of VSV vectors expressing the HIV Env gp160 and the simian immunodeficiency virus (SIV) Gag p55 elicited potent antigen-specific immune responses [59]. Moreover, vaccinated macaques were protected against AIDS. Other VSV-based HIV vaccine candidates such as rVSV-B6-NL4.3Env/SIVtm, rVSV-B6-NL4.3Env/Ebtm, and rVSV-B6-A74Env(PN6)/SIVtm, when administered to mice induced HIV gp140-specific antibody responses [60]. In the context of RABV, mice immunized with an RABV vector expressing the HIV-1 gp160 envelope protein elicited HIV-1 neutralizing antibodies after a boost injection with recombinant gp120/gp41 protein [61].
The annual global seasonal influenza virus threats have put pressure on vaccine development against influenza A virus and avian influenza virus strains. For instance, the HA protein of the highly pathogenic avian influenza virus (HPAIV) has been expressed from an MV vaccine strain (rMV-Ed-H5HA) or a wild-type MV-derived mutant (rMV-HL-Vko-H5HA) [62]. Immunization of cynomolgus monkeys elicited both MV- and HPAIV-specific antibodies. Furthermore, vaccinated monkeys showed less severe pneumonia, faster recovery from influenza symptoms, and suppression of viral shedding. Moreover, strong immune responses were observed in cotton rats immunized with an MV vector expressing the HA protein from the influenza A/Sapporo/107/2013 (H1N1pdm) strain [63]. Additionally, cotton rats immunized with MV AIK-C were protected against challenges with influenza A virus. In the case of VSV-based vaccine development, the VSV-G gene was replaced by the HPAIV HA gene, HPAIV A/FPV/Rostock/34 (H7N1), generating the replication-deficient VSV*DeltaG(HA) vector [64]. Intramuscular immunization of chickens resulted in high antibody titers, which neutralized avian influenza virus subtypes H7N1 and H7N7 but not H5N2. Moreover, immunized chickens were protected against challenges with lethal doses of the HPAIV A/chicken/Italy/445/99 (H7N1) strain. In another study, VSV vectors expressing the influenza HA and neuraminidase (NA) provided protection against lethal challenges in mice immunized at least 24 h after the virus challenge [65]. It was determined that HA-specific immune responses were essential for achieving full protection. Moreover, the engineering of a VSV vector expressing the H5 HA of the H5N1 HPAIV A/Vietnam/1203/04 (VN1203), and the NA of the mouse-adapted H1N1 influenza virus A/Puerto Rico/8/34 (PR8) generated a vaccine candidate, which was non-pathogenic in mice [66]. A single intramuscular or intranasal injection protected mice from lethal challenges with H5N1 influenza virus. A fast-acting VSV-based pan-H5 influenza virus vaccine was targeted by engineering different antigen designs [67]. Vaccination of mice confirmed that a single dose of VSV expressing the full-length HA gene (HAfl) provided 100% protection. The response was rapid as uniform protection was achieved when the vaccine candidate was administered only 3 days prior to the challenge. A single vaccination elicited cross-protective H5-specific antibodies and resulted in protection against challenges with different H5 clade 2 viruses, which is an indication of a pan-H5 influenza virus vaccine.
Without doubt, due to the current COVID-19 pandemic, vaccine development against coronaviruses has overshadowed any other vaccine initiative. So far, adenovirus vector-based SARS-CoV-2 vaccines have demonstrated good safety and efficacy in animal models and have provide more than 90% vaccine efficacy in human clinical phase I/IIa [68] and phase III trials [69]. At the end of 2020 and the beginning of 2021, these vaccine candidates received Emergency Use Authorization (EUA) in several countries allowing global mass vaccination to start [70, 71]. Issues related to new SARS-CoV-2 variants emerging and the discovery of rare cases of vaccine-induced immune thrombotic thrombocytopenia (VITT) [72] have added demands to develop new vaccines. In this context, attention has also been dedicated to negative stranded self-replicating RNA viruses. The Severe Acute Respiratory Syndrome (SARS) and Middle East Respiratory Syndrome (MERS) outbreaks prior to the COVID-19 pandemic triggered the development of vaccines against SARS-CoV and MERS-CoV, respectively. In the context of vaccine development against SARS, live-attenuated MV vectors expressing the SARS-CoV spike (S) protein and nucleocapsid (N) protein were evaluated in cell lines and in transgenic mice susceptible to MV infections [73]. The immunization elicited high antibody responses against MV and SARS-CoV S and N. The combination of MV-SARS-CoV-S and MV-SARS-CoV-N elicited similar antibody responses as seen for either MV construct alone. In another approach, a live attenuated MV was engineered to express the full-length membrane-anchored SARS-CoV S protein and its secreted soluble ectodomain (SARS-Ssol) [74]. Immunization of mice with MV-SARS-CoV-S elicited high titer neutralizing antibodies and provided full protection from intranasal challenges with SARS-CoV. Compared to immunization with an Ssol protein the immune responses from MV-SARS-CoV-S were stronger and Th1-biased. Related to VSV, an attenuated replication-competent VSV vector expressing the SARS-CoV S elicited SARS-neutralizing antibodies in immunized mice [75]. Moreover, mice showed long-term resistance to SARS-CoV challenges after a single vaccination. However, for safety reasons, a replication-defective, single cycle VSV vector was engineered by replacing the VSV G gene with the SARS-CoV S gene [76]. Intramuscular immunization of mice with the VSVΔG-SARS-CoV S vector induced significantly stronger antibody responses compared to replication-competent VSV vectors, which were 10 times higher than needed for protection against SARS-CoV
MV vectors have been engineered to express the full-length MERS-CoV S protein (MERS-S) and a truncated soluble variant (MERS-solS) [77]. Immunization of mice with MV-MERS-S and MV-MERS-solS elicited both MV- and MERS-CoV neutralizing antibodies and provided protection against MERS-CoV challenges. In another study, the replication-competent MV vector expressing the MERS-CoV S protein elicited robust neutralizing antibody titers in immunized mice [78]. In contrast, immunization with MV vectors expressing the MERS-CoV N protein did not induced MERS-specific antibody responses in vivo. VSV vectors have also been applied for the expression of the MERS-CoV S protein replacing the VSV-G protein [79]. Immunization of rhesus macaques generated robust Th1-biased antibody and T-cell responses after a single intramuscular or intranasal injection. RABV vectors have also been employed for MERS. An inactivated RABV vector was engineered to express the MERS-CoV full-length S protein [80]. However, the RABV-MERS-CoV S construct generated significantly reduced titers, which triggered the construction of an RBV vector expressing the MERS-CoV S1 domain fused to the RABV G protein C terminus [80]. Immunization of mice with RABV-MERS-CoV S1 resulted in protection against challenges with MERS-CoV.
In the context of SARS-CoV-2 and COVID-19 vaccine development MV vectors have been utilized for the expression of the SARS-CoV-2 S protein. Immunization of mice generated robust Th1-biased antibody and T-cell immune responses [81]. Based on preclinical findings the MV-SARS-CoV-2 S vaccine candidate TMV-083 was evaluated in a randomized, placebo-controlled phase I clinical trial [82]. However, due to disappointingly weak immune responses detected in vaccinated volunteers, the phase I study was terminated [83]. Both live and inactivated RABV expressing the SARS-CoV-2 S1 domain elicited potent virus-neutralizing antibodies in BALB/c mice at levels higher than observed in convalescent COVID-19 patients [84]. In another approach, VSV vectors were used for the expression of the SARS-CoV-2 S protein [85]. Neutralizing SARS-CoV-2-specific antibodies were induced in immunized mice. Moreover, protection against SARS-CoV-2 challenges was achieved. In another study, a replication-competent VSV vector, where the VSV G gene was replaced by the SARS-CoV-2 S gene, elicited strong antibody responses and protected immunized mice from SARS-CoV-2 challenges [86]. Moreover, mice expressing the human ACE2 receptor showed significantly reduced viral infection and inflammation in the lung, providing protection against pneumonia. In another approach, a methyltransferase-defective recombinant VSV vector was engineered for the expression of the full-length SARS-CoV-2 S, S1, and the receptor binding domain (RBD) [87]. The mtdVSV-SARS-CoV-2-S elicited robust SARS-CoV-2-specific neutralizing antibodies and Th1-biased T-cell immune responses in mice. Immunization of Syrian golden hamsters induced SARS-CoV-2-specific neutralizing antibodies of a higher magnitude than observed in convalescent COVID-19 patients and provided complete protection against SARS-CoV-2. VSV-based SARS-CoV-2 vaccine candidates have been subjected to clinical trials. For example, the VSV-SARS-CoV-2 S vaccine candidate V590 was evaluated in 252 volunteers in a phase I clinical trial [88]. Although the vaccination procedure was determined safe and well tolerated the inferior immune responses compared to levels in convalescent COVID-19 patients resulted in the termination of the trial [89]. Also, the application of VSVΔG-SARS-CoV-2 S has been planned for clinical evaluation. Healthy volunteers will receive a single dose of 5 × 105, 5 × 106, or 5 × 107 pfu of VSVΔG-SARS-CoV-2 in the first phase of a dose-escalation phase I/II study [90]. Furthermore, a phase II/III trial is planned [91].
Negative strand self-replicating RNA virus vectors and cancer
In addition to infectious diseases, negative strand self-replicating RNA viruses have been targeted for vaccine development for various cancers. In the case of cancers, the approaches have included not only vaccine development, but also cancer gene therapy and cancer immunotherapy. As for infectious diseases, vaccine development relies to a large part on overexpression of antigens, in this case, tumor antigens, for the induction of relevant antibody responses. In the context of cancer gene therapy, viral vectors have been used for the delivery of toxic or anti-tumor genes. Cancer immunotherapy involves the application of viral vectors as delivery vehicles for immunostimulatory genes such as interleukins and other cytokines. Oncolytic viruses have frequently been used in cancer therapy due to their specific replication in and killing of tumor cells without causing damage to normal cells [92]. Among negative strand self-replicating RNA viruses, attenuated MV strains [93] and engineered VSV vectors [94] are available. Examples of preclinical and clinical studies on cancer prevention and therapy using negative strand self-replicating RNA virus delivery vehicles are described below and summarized in Table 2.
The modest success in treating brain tumors has accelerated the application of viral vectors for cancer therapy and immunotherapy. More than 20 years ago it was demonstrated that VSV can selectively induce cytolysis of several human cell lines probably through apoptotic activity [95]. Moreover, VSV has demonstrated a potent inhibition of p53-null C6 glioblastoma tumors without affecting normal tissue. The replication-competent VSVrp30a strain expressing GFP was administered intravenously in mice showing rapid targeting and destruction of different types of human and mouse tumors including glioblastoma and mammary tumors [96]. Moreover, both tumors implanted in the brain and peripherally were infected by VSVrp30a-GFP. Intranasal administration resulted in selective infection and killing of olfactory bulb tumors without targeting normal cells. In another study, the attenuated VSV-p1-GFP variant showed killing of human U87 glioblastoma cells but caused no adverse neurological effects after systemic administration in mice [97]. A chimeric VSV vector expressing the CHIKV envelope polyprotein (E3-E2-6K-E1) in place of VSV G demonstrated selective infection and elimination of a broad range of human glioma cells [98]. In contrast to glioma cells, normal human astrocytes showed attenuated infection and viral replication was not detected. SCID mice implanted with human U87 glioma cells were intratumorally injected with VSVΔG-CHIKV, which resulted in substantial tumor regression and greatly enhanced survival. Moreover, the safety and antitumor effect of the G protein less fusion-associated small transmembrane VSV (GLESS-FAST-VSV) were evaluated in F344 rats and C57BL/6 mice [99]. GLESS-FAST-VSV infected and killed brain tumor cells with a significantly reduced toxic effect on normal cells. Multiple injections of GLESS-FAST-VSV in animals with implanted brain tumors provided a significantly prolonged survival showing little neurotoxicity. In the context of MV, oncolytic MV strains have shown promises in glioma treatment [100]. For instance, a single intratumoral administration of the oncolytic MV-GFP vector resulted in complete tumor regression and prolonged survival in mice [101]. Furthermore, a statistically significant increase in survival was discovered in an intracerebral mouse xenograft model treated with the oncolytic MV-GFP vector [102]. In another approach, GFP, carcinoembryonic antigen (CEA), and sodium iodide symporter (NIS), known for its therapeutic effect on cancers, were expressed from MV vectors [103]. Transduction of glioma stem cells (GSC) resulted in MV replication and cytotoxic activity. Implantation of MV-GFP transduced GSCs in the right caudate nucleus of nude mice resulted in significantly prolonged survival. In the context of clinical trials, a phase I study in patients with recurrent glioblastoma multiforme applying the MV-CEA vectors has been conducted although no results are available yet [104, 105].
In the context of breast cancer, the oncolytic VSV containing the M51R mutation in the matrix (M) protein gene has been applied for therapy of experimental breast cancer using VSV(M51R)-LacZ [106]. Intravenous administration of BALB/c mice implanted with 4T1 tumors demonstrated infection of multiple breast cancer lesions without causing lung toxicity, indicating safe and effective treatment of breast cancer metastases. In another study, recombinant VSV-MΔ51 expressing the cytosine deaminase/uracil phosphoribosyl transferase (CD-UPRT) suicide gene and 5-fluorocytosine (5-FC) prodrug showed the enhanced killing of human MCF-7 breast cancer cells [107]. Furthermore, treatment of mice bearing TSA mammary adenocarcinoma xenografts extended the survival time. In the case of MV, a recombinant vector was disenabled to use the signaling lymphocyte activation molecule (SLAM) targeting the poliovirus receptor-related 4 (PVRL4) and resulting in strong oncolytic activity in breast cancer cells [108]. The rMV-SLAMblind vector showed antitumor activity against human breast cancer xenografts in mice and the safety was evaluated in MV seronegative monkeys, which generated no clinical symptoms of MV. In another study, a live attenuated MV was evaluated in MCF-7 and CAL-51 breast cancer cell lines and in breast cancer cells derived from BRCA1/BRCA2-positive cancer patients [109]. MV effectively infected, induced apoptosis, and killed breast cancer cells suggesting the potential of safe and efficient anticancer treatment. Moreover, the attenuated MV Edmonston strain (MV-Edm) re-sensitized breast cancer cells to doxorubicin and ionizing radiation, which could provide a new strategy for the treatment of radio- and chemo-resistant breast cancer [110].
Cervical cancer can be considered as a special type of cancer as vaccine approaches have mainly aimed at providing protection and at a lesser extent therapeutic efficacy. The human papilloma virus (HPV) vaccine Gardasil, based on recombinant protein expression of the HPV L1 capsid protein in Saccharomyces cerevisiae, was approved by the FDA in 2006 [111]. Despite the success of Gardasil, there has been a demand on the development of new cervical cancer vaccines. In this context, MV vectors expressing the HPV16 L1 capsid protein showed strong humoral responses in immunized transgenic mice [112]. Moreover, the MV-HPV16 L1 vaccine was compared to HPV16 L1 and HPV18 L1 vaccines produced in Pichia pastoris [113]. Immunization of non-human primates elicited strong neutralizing antibody responses for both MV- and yeast-based vaccines. In the case of VSV, the E1, E2, E6, and E7 proteins of the cottontail rabbit papillomavirus (CRPV) were expressed from a VSV-vector and immunization of rabbits reduced the volume of papilloma significantly [114]. The VSV-E7 immunization was superior reducing tumor volumes by 96.9%, and also resulted in eradication of disease. Moreover, immunization of C57BL/6 mice with a VSV vector expressing HPV16 E7 elicited specific T cell responses and generated a 10-fold reduction in tumor volume [115].
Oncolytic MV vectors have been utilized for the expression of granulocyte macrophage-colony stimulating factor (GM-CSF) in a mouse adenocarcinoma MC38cea model [116]. The tumor progression was delayed after intratumoral MV-GM-CSF administration, which also prolonged the survival time of immunized animals. Tumor remission was complete in one-third of mice and tumor re-engraftment was further rejected. In another approach, oncolytic MV vectors expressing IL-12 and IL-15 were evaluated in MC38cea and B16hCD46 tumor models [117]. The MV-IL-15 vector enhanced T- and NK-cell filtration in both tumor models, but the MV-IL-12 vector generated more robust viral gene expression and immune activation, which resulted in superior anticancer activity. In another study, MV-based expression of IL-12 was evaluated in colon cancer cells and in rats [118]. MV-IL-12 administration in rats enhanced anti-tumor activity and immunity. In the context of an oncolytic VSV vector, expression of enhanced secreted IL-15 resulted in strong local expression in CT-26 colon tumors [119]. Immunization of mice enhanced both anti-tumoral T-cell responses and prolonged the survival time. In another study, an oncolytic VSV vector expressing the Newcastle disease virus (NDV) fusion protein mutant L289A downstream of the VSV G protein showed significantly enhanced survival in a rat liver metastasis model [120]. Moreover, four out of seven immunized rats demonstrated long-term survival exceeding 100 days. Although prolonged survival was also observed in the lung metastasis model, long-term survival was not achieved in any rat. In another study, the M protein mutant of VSV (M51R VSV) was evaluated in BALB/c mice with luciferase-expressing CT-26 peritoneal tumors for intraperitoneal tumor growth and overall survival [121]. A single intraperitoneal administration of 5 × 106 pfu of M51R VSV particles decreased luciferase bioluminescence and improved the survival of mice.
Related to lung cancer, the oncolytic MV Hu-191 strain showed significant tumor growth suppression and prolonged survival in C57BL/6 mice with Lewis lung carcinoma (LLC) [122]. In another study, the oncolytic MV Schwarz strain suppressed the growth of established lung and colorectal adenocarcinomas in nude mice with A549 and Caco-2 tumors [123]. Moreover, the MV-Edm strain was applied for the expression of CEA in non-small-cell lung cancer (NSCLC) cell lines and in transgenic mice [124]. MV-Edm-CEA induced apoptosis in NSCLC cell lines and tumor regression was observed after intratumoral administration in mice. In the context of VSV, expression of interferon-β (VSV-IFNβ) provided reduced tumor growth in mice with H2009 and A549 lung xenografts after intratumoral administration [125]. Moreover, tumor regression, prolonged survival, and cure were achieved in 30% of immunized mice bearing syngeneic LM2 lung tumors [125]. In another study, VSV-IFNβ based immunization was combined with ruxolitinib in an immunocompetent NSCLC mouse model, which resulted in improved survival of immunized mice [126]. The chimeric VSV-GP vector has been evaluated in syngeneic C57BL/6 and athymic nude mice implanted with subcutaneous LLC1 lung tumors [127]. Administration of VSV-GP demonstrated tumor-to-tumor spread of viral progeny in bilateral tumors and widespread infection and killing of tumor cells.
In the context of leukemia and lymphoma, clinical observations from the 1970s and 1980s indicated that regression was observed in patients infected with MV [128, 129]. In the case of leukemia, attenuated MV has been verified as a potential therapeutic agent for pediatric acute lymphoblastic leukemia (ALL) ex vivo in a panel of pediatric xenografted and naive primary ALLs and against four different ALL xenografts of B-lineage in SCID mice [130]. The MV effectively killed leukemia cells but spared normal human blood cells ex vivo. Intravenous injection of mice eradicated leukemia blasts in the hematopoietic system and provided long-term survival in three out of four xenografted B-lineage leukemias. In another approach, acute myeloid leukemia (AML) cell lines and primary AML cells from patients were infected with MV vectors expressing GFP or super cytosine deaminase (SCD) [131]. As SCD converts the prodrug 5-FU into 5-fluorouracil (5-FU) with chemotherapeutic activity, the combination therapy of MV and SCD substantially reduced the number and viability of leukemia cells by induction of apoptosis. Oncolytic VSV vectors have been evaluated in primary human T-lymphocytic virus type 1 (HTLV-1) infected T-lymphocytes from adult T-cell leukemia (ATL) patients [132]. VSV did neither replicate in HTLV-1 infected non-leukemic cells nor in naive CD4+ T-lymphocytes from healthy individuals. Although activation with anti-CD3/CD28 monoclonal antibodies induced limited VSV replication and oncolysis, it was four times higher in ATL cells. In another approach, intravenous administration of VSV-mIFNβ-NIS vectors demonstrated dose-dependent responses in syngeneic AML CL1498 tumors [133]. Combination therapy with VSV-mIFNβ-NIS and anti-PD-L1 antibodies enhanced antitumor activity and significantly prolonged survival. In attempts to compensate for the overexpression of anti-apoptotic B-cell leukemia/lymphoma 2 (BCL-2) interfering with apoptosis, oncolytic VSV vectors combined with the BCL-2 inhibitor obatoclax, synergistically induced cell death in primary chronic lymphocytic leukemia (CLL) cells and reduced tumor growth in SCID mice with implanted A20 lymphoma tumors [134].
Intratumoral administration of the live attenuated MV-Edm strain with or without the LacZ reporter gene induced regression of large human lymphoma xenografts in SCID mice [135]. Furthermore, intravenous administration generated significant decrease in DoHH2 and Raji tumor progression and MV replication was detected in residual tumors. In another approach, MV-based expression of NIS was applied against mantle cell lymphoma (MCL), an aggressive but radiosensitive form of B-cell non-Hodgkin’s lymphoma [136]. In mice, infection of metastases was faster and more intense than for primary tumors. Combination therapy of MV-NIS and systemic iodine-131 showed more rapid tumor regression than either therapy alone. Interestingly, only SLAM-based entry of MV provided efficient virus spread, tumor regression, and extended survival in mice. In the context of clinical trials, the MV-Edmonston-Zagreb strain was intratumorally administered to five patients with cutaneous T-cell lymphomas (CTCLs) in a phase I clinical trial [137]. The treatment was safe and well tolerated, which generated clinical responses. Moreover, patients with classical Hodgkin lymphoma (cHL) been subjected to treatment with oncolytic MV and VSV [138]. In this context, a CD30-targeted MV vector (MV-CD30) and a VSV-vector where the VSV-G was replaced by the CD30-targetd MV glycoproteins (VSV-CD30) were engineered. Both MV-CD30 and VSV-CD30 selectively destroyed CD30+-positive cHL tumor cells although VSV-CD30 generated much higher titers and more rapidly and efficiently killed cHL-derived cell lines. Intratumoral and systemic administration of VSV-CD30 significantly decreased the tumor growth and prolonged survival in mice with implanted cHL xenografts.
In the case of melanoma, an oncolytic MV virus carrying the melanoma-associated antigen (HMWMAA) demonstrated highly specific infection and spread in melanoma cells [139]. The therapeutic efficacy was further improved by the introduction of the bifunctional fusion suicide (FCU1) gene, which is coding for the yeast-derived prodrug convertases CD and uracil phosphoribosyl transferase. Combination therapy of MV-FCU1-αHMWMAA and the 5-FC prodrug converted to 5-FU generated extensive cytotoxicity in melanoma cells and in non-infected cells through excessive bystander killing. Furthermore, biopsies of melanoma skin metastases revealed targeting by the MV-FCU1-αHMWMAA vector. An important issue related to the treatment of melanoma with oncolytic MV vectors has been to determine what renders human melanoma cells susceptible to MV infections [140]. It was demonstrated both in vitro and in vivo that the IFN pathway was fully functional in MV resistant cells, and sensitive cells pre-treated with IFN showed resistance against MV infections. Moreover, treatment with the IFN pathway inhibitor ruxolitinib rendered cells susceptible to MV infections. The oncolytic potential of the MV Leningrad-16 (L-16) strain was evaluated in human metastatic melanoma cell lines and the melZ mouse melanoma model [141]. MV-L-16 replicated efficiently in melanoma cell lines resulting in direct killing of tumor cells. Furthermore, significant tumor growth inhibition was achieved in the melanoma mouse model. In the context of VSV, subcutaneous administration of oncolytic VSV into B16ova melanomas resulted in tumor regression but no viral replicative burst in C57BL/6 mice [142]. However, intratumoral administration induced acute proinflammatory reactions and virus clearance. It was further demonstrated that a single cycle of oncolytic VSV was as efficient as a fully replication-competent VSV. In another study, the VSV-G protein was relaced by the non-neurotropic lymphocytic choriomeningitis virus (LCMV) glycoprotein (GP) in the VSV vector [143]. Evaluation in subcutaneous A375 and B16-OVA syngeneic mouse tumor models demonstrated significant tumor regression and prolonged survival of immunized mice. Among rhabdoviruses, in addition to VSV, Maraba virus has demonstrated oncolytic potential. The Maraba MG1 strain was engineered to express human dopachrome tautomerase (hDCT), but immunization with MG1-hDCT was insufficient for induction of antitumor immunity and for providing therapeutic efficacy in mice with implanted B16-F10 metastases [144]. However, Maraba MG1 showed potency for boosting DCT-specific immune responses after priming with an adenovirus expressing hDCT.
In the context of ovarian cancer, the MV-Edm strain expressing CEA showed oncolytic activity in ovarian tumor cells, but only minimal cytotoxicity in normal ovarian surface epithelial and mesothelial cells [145]. Intratumoral injection into SKOV3ip.1 human ovarian epithelial xenografts resulted in complete regression of 80% of tumors in athymic mice. Moreover, intraperitoneal administration extended the survival of mice with advanced SKOV3ip.1 tumors by more than 50 days. MV vectors have also been subjected to engineering for tumor-specific targeting by the introduction of a single-chain antibody (scFV) sequence for the alpha-folate receptor (αFR) [146]. Reduction in tumor volume and extended survival was observed in mice implanted with SKOV3ip.1 ovarian xenografts after intratumoral administration. In a dual therapy approach, MV vectors expressing CEA and NIS were administered to mice implanted with SKOV3ip.1 ovarian tumor xenografts [147]. The combination therapy was superior to treatment with either MV-CEA or MV-NIS alone. The MV-CEA vector was subjected to a phase I clinical study in patients with taxol and platinum-refractory recurrent ovarian cancer [148]. Intraperitoneal MV-CEA administration was safe, well tolerated, and demonstrated dose-dependent biological activity in heavily pre-treated patients. Stable disease (SD) was observed in 14 out of 21 patients. In the case of VSV, recombinant plasmid DNA carrying a VSV matrix protein (MP) liposome complex transfected into SKOV3 ovarian cancer cells induced apoptosis and intraperitoneal administration reduced tumor weight by 90% [149]. In another study, the VSV MP liposome complex was administered to nude mice with implanted A2780s and A2780cp ovarian tumors [150]. The treatment resulted in 87–98% inhibition of A280s and A2780cp tumor xenografts and extended the survival time in mice. In a combination therapy approach, encapsulation of paclitaxel in DPP particles enhanced the VSV MP expression, which induced antiproliferation and apoptosis in SKOV3 ovarian tumor cells [151]. Intraperitoneal administration demonstrated efficient inhibition of metastases, inhibition of tumor cell proliferation, and suppression of tumor angiogenesis. The VSV-LCMV GP vector showed oncolytic activity in several ovarian cancer cell lines [152]. Moreover, administration of VSV-LCMV GP into subcutaneous and orthotopic A2780 ovarian cancer mouse models led to tumor regression, which was further enhanced when combined with administration of the JAK1/2 inhibitor ruxolitinib.
Pancreatic cancer has been targeted by immunization of SCID mice with implanted KLM1 and Capan-2 pancreatic tumor xenografts with MV-SLAMBlind vectors [153]. Intratumoral injection resulted in significant suppression of tumor growth. Furthermore, the oncolytic MV was subjected to combination therapy with gemcitabine in human MIA Paca-2, PANC-1, and BxPC-3 pancreatic cancer cell lines [154]. The combination treatment reduced the tumor cell mass of more than 50% and gemcitabine only slightly diminished viral replication in tumor cells. VSV vectors have been applied for therapeutic efficacy against the highly aggressive pancreatic ductal adenocarcinoma (PDAC) [155]. The oncolytic activity of VSV was superior in comparison to Sendai virus and respiratory syncytial virus (RSV) despite some heterogeneity of PDAC cells toward VSV susceptibility. Moreover, wild-type VSV, VSV-GFP, and VSV-ΔM51-GFP showed oncolytic activity in five PDAC cell lines [156]. In vivo, administration of VSV-ΔM51-GFP in mice implanted with PDAC xenografts resulted in significant decrease in tumor growth, which was further improved by co-administration of gemcitabine. In another study, the cause of resistance or susceptibility of PDAC cells to VSV was investigated showing constitutive co-expression of interferon-stimulated genes (ISGs) such as MX1, EPSTI1, XAF1, and GBP1 in VSV-resistant, but not in VSV-permissive PDAC cells [157]. It was also demonstrated that ruxolitinib can down-regulate ISG expression and short hairpin RNA (shRNA) based inhibition of MX1 supported VSV-ΔM51-GFP replication in VSV-resistant PDAC cells. In another study, a VSV vector was engineered where the VSV-G was replaced by the MV envelope proteins F and H [158]. The VSV-HF vector was evaluated both in vitro and in vivo in models for hepatobiliary and pancreatic cancers. A single intratumoral administration of VSV-HF demonstrated complete tumor regression and extended survival in mice with implanted Hep3B hepatocellular carcinoma. In contrast, this effect was not observed after immunization of mice with BxPC-3 pancreatic cancer xenografts.
In the context of prostate cancer, intratumoral administration of MV-CEA in mice with implanted prostate PC-3 xenografts substantially delayed tumor growth and prolonged survival [159]. In another approach, a single-chain antibody (sc-Fv) specific for the extracellular domain of the prostate-specific membrane antigen (PSMA) was introduced into the MV vector [160]. The therapeutic efficacy was evaluated in mice with implanted LNCaP and PC3-PSMA prostate tumor xenografts. The infection and cytopathic killing occurred exclusively in PSMA-positive prostate cancer cells. Radiation therapy further enhanced the therapeutic efficacy. The co-administration of oncolytic MV and mumps virus (MuV) vectors in mice with implanted PC-3 prostate xenografts resulted in superior anti-tumor activity and extended survival compared to administration of either MV or MuV alone [161]. Related to VSV, infection of human DU145 and PC3 prostate cell lines with the VSV-ΔM51-GFP vector resulted in efficient VSV replication, apoptosis, and tumor cell killing [162]. Eradication of tumor cells was demonstrated in nude mice immunized with VSV-ΔM51-GFP, which prolonged the survival time. In contrast, normal tissue was relatively unaffected. The previously described VSV-LCMV GP vector [152] has also been subjected to prostate cancer therapy. VSV-LCMV GP efficiently infected six out of seven prostate cancer cell lines [163]. Moreover, long-term remission was achieved in the DU145 prostate cancer tumor model after intratumoral immunization of BALB/c Rag2−/−γc−/− mice. Long-term cure was also detected after intravenous treatment of subcutaneous tumors and bone metastases. Finally, the effect of the combination of VSV with natural agents with anti-tumor properties was investigated in vitro and in vivo [164]. The VSV-induced oncolysis was enhanced after pre-treatment with curcumin in the PC-3 prostate cancer cell line and in a prostate cancer mouse model.
Conclusions
In summary, self-replicating negative strand RNA viruses have demonstrated both prophylactic and therapeutic efficacy in various animal models for infectious diseases and different cancers. Although self-replicating positive strand RNA viruses such alphaviruses and flaviviruses have also shown promising results in vaccine development and cancer therapy and possess a certain advantage of direct translation of their genomic RNA in the cytoplasm of infected host cells, both measles viruses and rhabdoviruses have proved their potential. Especially, VSV-based vaccines have been highly successful, which has been demonstrated by the approval of vaccines against EVD by both the FDA and the EMA [47]. However, recent vaccine development against SARS-CoV-2 has served as a reminder of the difficulty of transferring vaccine efficacy from animal models to human applications as clinical trials for both measles virus [82, 83] and VSV [88, 89] were prematurely terminated due to only weak immune responses in vaccinated volunteers. In the area of cancer vaccines and cancer immunotherapy, a large number in animal models have demonstrated tumor regression, eradication of tumors, and even long-term remission. However, a relatively small number of clinical trials have been conducted, so far. For this reason, it is essential to be able to broaden the repertoire of clinical evaluations in the future.
How can progress be achieved in the future? Obviously, there is no magic bullet or a specific viral vector that has been shown to be universally applicable and providing superior efficacy for either vaccine development or cancer therapy. Oncolytic viruses have become attractive due to their capacity to specifically infect and replicate in tumor cells, furthermore, killing the host cells while leaving normal tissue unaffected. Much attention has also been paid to combinatorial approaches. Several studies have demonstrated improved efficacy of combining viral vectors with antiviral and cancer drugs, respectively. Even pre-treatment with natural agents such as curcumin has resulted in enhanced oncolysis of VSV vectors. Moreover, the combination of viral delivery vehicles with radiotherapy has demonstrated improvement in cancer therapy.
Plenty of attention has also been dedicated to vector engineering to design safer and more efficient delivery systems. Efforts have been made to identify mechanisms and pathways facilitating the targeting and infection of appropriate cells, in the case of vaccines antigen-producing cells and for cancer therapy tumor cells. Especially for cancer therapy it has been essential not to cause damage to normal tissue. As has been learnt from clinical evaluation of vaccine candidates, the route of administration, single or prime-boost administration, and dosage are important issues to address. The COVID-19 pandemic has also taught us how vaccine development can be accelerated through fruitful collaboration between academic research institutions, pharmaceutical companies, and governmental organizations. It has demonstrated the importance of being prepared for emerging viruses and the necessity to deal with novel mutants/variants to eradicate the pandemic. For these efforts and for future global well-being self-replicating negative strand RNA viruses can play an important role.
References
Lundstrom K. Viral vectors in gene therapy. Diseases. 2018;6:42.
Bloom K, van der Berg F, Arbuthnot P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 2021;28:117–29.
Samulski R, Muzycka N. AAV-mediated gene therapy for research and therapeutic purposes. Annu Rev Virol. 2014;1:427–51.
Vigna E, Naldini L. Lentiviral vectors: Excellent tools for experimental gene transfer and promising candidates for gene therapy. J Gen Med. 2000;2:308–16.
Strauss JH, Strauss EG. The alphaviruses: Gene expression, replication, and evolution. Microbiol Rev. 1994;58:491–562.
Frolov I, Hoffman TA, Pragal BM, Dryga SA, Huang H, Schlesinger S, et al. Alphavirus-based expression vectors: Strategies and applications. Proc Natl Acad Sci USA. 1996;93:11371–7.
Liljestrom P, Garoff H. A new generation of animal cell expression vectors based on the Semliki Forest virus replicon. Biotechnology. 1991;9:1356–61.
Xiong C, Levis R, Shen P, Schlesinger S, Rice CM, Huang HV. Sindbis virus: An efficient, broad host range vector for gene expression in animal cells. Science. 1989;243:1188–91.
Davis NL, Willis LV, Smith JF, Johnston RF. In vitro synthesis of infectious Venezuelan equine encephalitis virus RNA from a cDNA clone: Analysis of a viable deletion mutant. Virology. 1989;171:189–204.
Lundstrom K. Self-amplifying RNA viruses as RNA vaccines. Int J Mol Sci. 2020;21:5130.
Walpita P, Flick R. Reverse genetics of negative-stranded RNA viruses: A global perspective. FEMS Microbiol Lett. 2005;244:9–18.
Whelan SPJ, Barr JN, Wetz GW. Transcription and Replication of Negative-Strand RNA Viruses. In: Kawoka Y, editor. Biology of negative stranded RNA viruses: The power of reverse genetics. Berlin, Heidelberg, New York: Springer Verlag; 2004. p. 61–119.
Radecke F, Spielhofer P, Schneider H, Kaelin K, Huber M, Dötsch C, et al. Rescue of measles viruses from cloned DNA. EMBO J. 1995;14:5773–84.
Mühlebach MD. Vaccine platform recombinant measles virus. Virus Genes. 2017;53:733–40.
Schneider H, Spielhofer P, Kaelin K, Dötsch C, Radecke F, Sutter G, et al. Rescue of measles virus using a replication-deficient vaccinia-T7 vector. J Virol Methods. 1997;64:57–64.
Martin A, Staeheli P, Schneider U. RNA polymerase II-controlled expression of antigenomic RNA enhances the rescue efficacies of two different members of the Mononegavirales independently of the site of viral genome replication. J Virol. 2006;80:5708–15.
Schnell MJ, Buonocore L, Kretzschmar E, Johnson E, Rose JK. Foreign glycoproteins expressed from recombinant vesicular stomatitis viruses are incorporated efficiently into virus particles. Proc Natl Acad Sci USA. 1996;93:11359–65.
Tani H, Morikawa S, Matsuura Y. Development and applications of VSV vectors based on cell tropism. Front Microbiol. 2012;2:272.
Hastie E, Grdzelishvili VZ. Vesicular stomatitis virus as a flexible platform for oncolytic virotherapy against cancer. J Gen Virol. 2012;93:2529–45.
Harty RN, Brown ME, Hayes FP, Wright NT, Schnell MJ. Vaccinia virus-free recovery of vesicular stomatitis virus. J Mol Microbiol Biotechnol. 2001;3:513–7.
Poli JG, Zhang L, Bridle BW, Stephenson KB, Rességuir J, Hanson S, et al. Maraba virus as a potent oncolytic vaccine vector. Mol Ther. 2014;22:420–9.
Ito N, Takayama-Ito M, Yamada K, Hosokawa J, Sugiyama M, Minamoto N. Improved recovery of rabies virus from cloned cDNA using a vaccinia virus-free reverse genetics system. Microbiol Immunol. 2003;47:613–7.
Osakada F, Callaway EM. Design and generation of recombinant rabies virus vectors. Nat Protoc. 2013;8:1583–601.
Ohara S, Inoue K, Yamada M, Yamawaki T, Koganezawa N, Tsuttsui K, et al. Dual transneural tracing in the rat entorhoinal-hippocampal circuit by intracerebral injection of recombinant rabies virus vectors. Front Neuroanat. 2009;3:1–11.
Kelvin AA. Outbreak of Chikungunya in the Republic of Congo and the global picture. J Infect Dev Ctries. 2011;5:441–4.
Jansen KA. The 2005-7 Chikungunya epidemic in Reunion: Ambiguous etiologies, memories, and meaning-making. Med Anthropol. 2013;32:174–89.
Chattopadhyay A, Aquilar PV, Bopp NE, Yarovinsky TO, Weaver SC, Rose JK. A recombinant virus vaccine that protects both against Chikungunya and Zika virus infections. Vaccine. 2018;36:3894–3900.
Ramsauer K, Tangy F. Chikungunya virus vaccines: Viral vector-based approaches. J Infect Dis. 2016;214:S500–S505.
Brandler S, Ruffie C, Combredet C, Brault J-B, Najburg V, Prevost M-C, et al. A recombinant measles vaccine expressing chikungunya virus-like particles is strongly immunogenic and protects mice from lethal challenge with chikungunya virus. Vaccine. 2013;31:3718–25.
Rossi SL, Comer JE, Wang E, Azar SR, Lawrence WS, Plante JA, et al. Immunogenicity and efficacy of a measles virus-vectored chikungunya vaccine in nonhuman primates. J Infect Dis. 2019;220:735–42.
Ramsauer K, Schwameis M, Firbas C, Mullner M, Putnak RJ, Thomas SJ, et al. Immunogenicity, safety, and tolerability of a recombinant measles virus-based chikungunya vaccine: a randomised, double-blind, placebo controlled, active-comparator, first-in-man trial. Lancet Infect Dis. 2015;15:519–27.
Reisinger EC, Tschismarov R, Beubler E, Wiedermann U, Firbas C, Loebermann M, et al. Immunogenicity, safety, and tolerability of the measles-vectored chikungunya virus vaccine MV-CHIK: a double-blind, randomised, placebo-controlled and active-controlled phase 2 trial. Lancet. 2019;392:2718–27.
Nasar F, Matassov D, Seymour RL, Latham T, Gorchakov RV, Novak RM, et al. Recombinant Isfahan virus and vesicular stomatitis virus vaccine vectors provide durable, multivalent, single-dose protection against lethal alphavirus challenges. J Virol. 2017;91:e01729–16.
Safronetz D, Mire C, Rosenke K, Feldmann F, Haddock E, Geissbert T, et al. A recombinant vesicular stomatitis virus-based Lassa fever vaccine protects guinea pigs and macaques against challenge with geographically and genetically distinct Lassa viruses. PLoS Negl Trop Dis. 2015;9:e0003736.
Stein DR, Warner BM, Soule G, Tierney K, Frost KL, Booth S, et al. A recombinant vesicular stomatitis-based Lassa fever vaccine elicits rapid and long-term protection from lethal Lassa virus infection in guinea pigs. NPJ Vaccines. 2019;4:8.
Banadyga L, Stein DR, Qiu X, Safronetz D. Pre-clinical development of a vaccine against Lassa fever. Can Commun Dis Rep. 2018;44:139–47.
Abreu-Mota T, Hagen KR, Cooper K, Jahrling PB, Tan G, Wirblich C, et al. Non-neutralizing antibodies elicited by recombinant Lassa-Rabies vaccine are critical for protection against Lassa fever. Nat Commun. 2018;9:4223.
Mateo M, Reynard S, Carnec X, Journeaux A, Baillet N, Schaeffer J, et al. Vaccines inducing immunity to Lassa fever glycoprotein and nucleoprotein protect macaques after a single shot. Sci Transl Med. 2019;11:eaaw3163.
A Trial to Evaluate the Optimal Dose of MV-LASV. ClinicalTrials.gov NCT04055454. https://clinicaltrials.gov/ct2/show/NCT04055454 (Accessed on August 26, 2021).
Shuai L, Wang X, Wen Z, Ge J, Wang J, Zhao D, et al. Genetically modified rabies virus-vectored Ebola virus disease vaccines are safe and induce efficacious immune responses in mice and dogs. Antivir Res. 2017;146:36–44.
Marzi A, Robertson SJ, Haddock E, Feldmann F, Hanley PW, Scott D-P, et al. VSV-EBOV rapidly protects macaques against infection with the 2014/2015 Ebola virus outbreak strain. Science. 2015;349:739–42.
Marzi A, Reynolds P, Mercado-Hernandez R, Callison J, Feldmann F, Rosenke R, et al. Single low-dose VSV-EBOV vaccination protects cynomolgus macaques from lethal Ebola challenge. EBioMedicine. 2019;49:223–31.
Jones SM, Stroher U, Fernando L, Qiu X, Alimonti J, Melito P, et al. Assessment of a vesicular stomatitis virus-based vaccine by use of the mouse model of Ebola virus hemorrhagic fever. J Infect Dis. 2007;196:S404–412.
Jones SM, Feldmann H, Stroher U, Geisbert JB, Fernando L, Grolla A, et al. Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses. Nat Med. 2005;11:786–90.
Henao-Restrepo AM, Longini IM, Egger M, Dean NE, Edmunds WJ, Camacho A, et al. Efficacy and effectiveness of an rVSV-vectored vaccine expressing Ebola surface glycoprotein: Interim results from the Guinea ring vaccination cluster-randomised trial. Lancet. 2015;386:857–66.
Henao-Restrepo AM, Camacho A, Longini IM, Watson CH, Edmunds WJ, Egger M, et al. Efficacy and effectiveness of an rVSV-vectored vaccine in preventing Ebola virus disease: Final results from the Guinea ring vaccination, open-label, cluster-randomised trial (Ebola Ca Suffit!). Lancet. 2017;389:505–18.
Maxmen, A. Ebola vaccine approved for use in ongoing outbreak. Nature. 2017. https://doi.org/10.1038/nature.2017.22024.
Hu HM, Chen HW, Hsiao Y, Wu SH, Chung HH, Hsieh CH, et al. The successful induction of T-cell and antibody responses by a recombinant measles virus-vectored tetravalent dengue vaccine provides partial protection against dengue-2 infection. Hum Vaccine Immunother. 2016;12:1678–89.
Lauretti F, Chattopadhyay A, de Oliveira França RF, Castro-Jorge L, Rose J, da Fonseca BAL. Recombinant vesicular stomatitis virus-based dengue-2 vaccine candidate induces humoral response and protects mice against lethal infection. Hum Vaccine Immunother. 2016;12:2327–33.
Jin H, Jiao C, Cao Z, Huang P, Chi H45, Bai Y, et al. An inactivated recombinant rabies virus displaying the Zika virus prM-E induces protective immunity against both pathogens. PLoS Negl Trop Dis. 2021;15:e0009484.
Kurup D, Wirblich, C, Schnell, MJ. Measles-based Zika vaccine induces long-term immunity and requires NS1 antibodies to protect the female reproductive tract in the mouse model of Zika. Preprint at bioRxiv 2020. https://doi.org/10.1101/2020.09.17.301622.
Zika-Vaccine Dose Finding Study Regarding Safety, Immunogenicity and Tolerability (V186-001). ClinicalTrials.gov NCT02996890. https://cliniclatriasl.gov/ct2/show/NCT02996890 (Accessed on August 31, 2021).
Safety and Immunogenicity of a Novel Vaccine Formulation MV-ZIKA-RSP (V187-001) (MV-ZIKA-RSP) NCT04033068. https://clinicaltrials.gov/ct2/show/NCT04033068 (Accessed on August 31, 2021).
Del Valle JR, Devaux P, Hodge G, Wegner NJ, McChesney MB, Cattaneo R. A vectored measles virus induces hepatitis B surface antigen antibodies while protecting macaques against virus challenge. J Virol. 2007;81:10597–605.
Cobleigh MA, Buonocore L, Uprichard SL, Rose JK, Robek MD. A vesicular stomatitis virus-based hepatitis B virus vaccine vector provides protection against challenge in a single dose. J Virol. 2010;84:7513–22.
Lorin C, Mollet L, Delebecque F, Combredet C, Hurtrel B, Charneau P, et al. A single injection of recombinant measles virus vaccine expressing human immunodeficiency virus (HIV) type 1 clade B envelope glycoproteins induces neutralizing antibodies and cellular immune responses to HIV. J Virol. 2004;78:146–57.
Liniger M, Zuniga A, Azzouz Morin TN, Combardiere B, Marty R, Wiegand M, et al. Recombinant measles viruses expressing single or multiple antigens of human immunodeficiency virus (HIV-1) induce cellular and humoral immune responses. Vaccine. 2009;27:3299–305.
Clarke DK, Cooper D, Egan MA, Hendry RM, Parks CL, Udem SA. Recombinant vesicular stomatitis virus as an HIV vaccine vector. Springer Semin Immunopathol. 2006;28:239–53.
Rose NF, Marx PA, Luckay A, Nixon DF, Moretto WJ, Donahoe SM, et al. An effective AIDS vaccine based on live attenuated vesicular stomatitis virus recombinants. Cell. 2001;106:539.
Mangion M, Gélinas J-F, Zadeh Gashti AB, Azizi H, Kiesslich S, Nassoury N, et al. Evaluation of novel HIV vaccine candidates using recombinant vesicular stomatitis virus vector produced in serum-free Vero cell cultures. Vaccine. 2020;38:7949–55.
Schnell MJ, Foley HD, Siler CA, McGettigan JP, Dietzschold B, Pomerantz RJ. Recombinant rabies virus as potential live-viral vaccines for HIV-1. Proc Natl Acad Sci USA. 2000;97:3544–9.
Fujiyuki T, Horie R, Yoneda M, Kuraishi T, Yasui F, Kwon H-J, et al. Efficacy of recombinant measles virus expressing highly pathogenic avian influenza virus (HPAIV) antigen against HPAIV infection in monkeys. Sci Rep. 2017;7:12017.
Ito T, Kumagai T, Yamaji Y, Sawada A, Nakayama T. Recombinant Measles AIK-C vaccine strain expressing influenza HA protein. Vaccines. 2020;8:149.
Kalhoro NH, Veits J, Rautenschlein S, Zimmer G. A recombinant vesicular stomatitis virus replicon vaccine protects chickens from highly pathogenic avian influenza virus (H7N1). Vaccine. 2009;27:1174–83.
Barefoot BE, Athearn B, Sample CJ, Ramsburg EA. Intramuscular immunization with a vesicular stomatitis virus recombinant expressing the influenza hemagglutinin provides post-exposure protection against lethal influenza challenge. Vaccine. 2009;28:79–89.
Ryder AB, Buonocore L, Vogel L, Nachbagauer R, Krammer F, Rose JK. A viable recombinant rhabdovirus lacking its glycoprotein gene and expressing influenza virus hemagglutinin and neuraminidase is a potent influenza vaccine. J Virol. 2015;89:2820–30.
Furuyama W, Reynolds P, Haddock E, Meade-White K, Le MQ, Kawaoka Y, et al. a single dose of a vesicular stomatitis virus-based influenza vaccine confers rapid protection against H5 viruses from different clades. NPJ Vaccines. 2020;5:4.
Sadoff J, Le Gars M, Shukarev G, Heerwegh, Truyers DC, de Groot AM, et al. Interim Results of a Phase 1-2a Trial of Ad26.COV2.S Covid-19 Vaccine. N Engl J Med. 2021;384:1824–35.
Voysey M, Sue Costa Clemens SA, Madhi SA, Weckx LY, Folegatti PM, Aley PK, et al. Safety and efficacy of the AdChOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised trials in Brazil, South Africa and the UK. Lancet. 2021;397:99–111.
Regulatory Approval of COVID-19 Vaccine AstraZeneca—GOV.UK. www.gov.uk (Accessed on September 2, 2021).
Ad26.COV2-S FDA Approval Status. drugs.com/history/ad26-cov2-s.html (Accessed on September 2, 2021).
Lundstrom K, Barh D, Uhal BD, Takayama K, Aljabali AAA, Mohamed Abd El-Aziz T, et al. COVID-19 vaccines and thrombosis—roadblock of dead-end street? Biomolecules. 2021;11:1020.
Liniger M, Zuniga A, Tamin A, Azzouz-Morin TN, Knuchel M, Marty RR, et al. Induction of neutralising antibodies and cellular immune responses against SARS coronavirus by recombinant measles virus. Vaccine. 2008;26:2164–74.
Escriou N, Callendret B, Lorin V, Combredet C, Marianneau P, Février M, et al. Protection from SARS coronavirus conferred by live measles vaccine expressing spike glycoprotein. Virology. 2014;452-3:32–41.
Kapadia SU, Rose JK, Lamirande E, Vogel L, Subbarao K, Roberts A. Long-term protection from SARS coronavirus infection conferred by a single immunization with an attenuated VSV-based vaccine. Virology. 2005;340:174–82.
Kapadia SU, Simon ID, Rose JK. SARS vaccine based on a replication-defective recombinant vesicular stomatitis virus is more potent than one based on a replication-competent vector. Virology. 2008;376:165–72.
Malczyk AH, Kupke A, Prüfer S, Scheuplein VA, Hutzler S, Kreuz D, et al. A highly immunigenic and protective middle east respiratory syndrome coronavirus vaccine based on a recombinant measles virus vaccine platform. J Virol. 2015;89:11654–67.
Bodmer BS, Fiedler AH, Hanauer JRH, Prüfer S, Mühlebach MD. Live-attenuated bivalent measles virus-derived vaccines targeting Middle East respiratory syndrome coronavirus induce robust and multifunctional T cell responses against both viruses in an appropriate moues model. Virology. 2018;521:99–107.
Liu R, Wang J, Shao Y, Wang X, Zhang H, Shuai L, et al. A recombinant VSV-vectored MERS-CoV vaccine induces neutralizing antibody and T cell responses in rhesus monkeys after single dose immunization. Antivir Res. 2018;150:30–38.
Wirblich C, Coleman CM, Kuryp D, Abraham TS, Bernbaum JG, Jahrling PB, et al. One.Health: a safe, efficient, dual-use vaccine for humans and animals against middle east respiratory syndrome coronavirus and rabies virus. J Virol. 2017;91:e02040–16.
Hörner C, Schürmann C, Auste A, Ebenig A, Muraleedharan S, Dinnon KH 3rd, et al. A highly immunogenic and effective measles virus-based Th1-biased COVID-19 Vaccine. Proc Natl Acad Sci USA. 2020;117:32657–66.
Clinical Trial to Evaluate the Safety and Immunogenicity of the COVID-19 Vaccine (COVID-19-101). https://clinicaltrials.gov/ct2/show/NCT04497298 (Accessed on September 2, 2021).
Merck Discontinues Development of SARS-CoV-2/COVID-19 Vaccine Candidates; Continues Development of Two Investigational Therapeutic Candidates. www.merck.com/news/merck-discontinues-development-of-sars-cov-2-covid-19-vaccine-candidates-continues-development-of-two-investigational-therapeutic-candidates/ (Accessed on September 2, 2021).
Kuryp D, Wirblich C, Ramage H, Schnell MJ. Rabies virus-based COVID-19 vaccine CORAVAX™ induces high levels of neutralizing antibodies against SARS-CoV-2. NPJ Vaccines. 2020;5:98.
Brett JB, Rothlauf PW, Chen RE, Kafai NM, Fox JM, Smith BK, et al. Replication-competent vesicular stomatitis virus vaccine vector protects against SARS-CoV-2-mediated pathogenesis in mice. Cell Host Microbe. 2020;28:465–74.
Case JB, Rothlauf PW, Chen RE, Kafai NM, Fox JM, Smith BK, et al. Replication-competent vesicular stomatitis virus vaccine vector protects against SARS-CoV-2-mediated pathogenesis in Mice. Cell Host Microbe. 2020;28:465–474e4.
Lu M, Zhang Y, Dravid P, Li A, Zeng C, Mahesh KC, et al. A methyltransferase-defective VSV-based SARS-CoV-2 vaccine candidate provides complete protection against SARS-CoV-2 infection in hamsters. J Virol. 2021. https://doi.org/10.1128/JVI.00592-21.
Dose Ranging Trial to Assess Safety and Immunogenicity of V590 (COVID-19 Vaccine) in Healthy Adults (V590-001). ClinicalTrials.gov NCT04569786. https;//clinicaltrials.gov/ct2/show/NCT04569786 (Accessed on September 2, 2021).
Merck and IAVI Discontinue Development of COVID-19 Vaccine Candidate V590. www.iavi.org/newsresources/press-releases/2021/merck-and-iavi-discontinue-development-of-covid-19-vaccine-candidate-v590 (Accessed on September 2, 2021).
Evaluate the Safety, Immunogenicity, and Potential Efficacy of an rVSV-SARS-CoV-2-S Vaccine. https://clinicaltrials.gov/ct2/show/NCT04608305 (Accessed on September 2, 2021).
Phase 2b Dose-confirmatory Trial to Evaluate the Safety, Immunogenicity and Potential Efficacy of an VSV-ΔG SARS-CoV-2 Vaccine (BRILIFE001) https://clinicaltrials.gov/ct2/show/NCT04990466 (Accessed on September 2, 2021).
Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016;107:1373–9.
Aref S, Bailey K, Fielding A. Measles to the rescue: A review of oncolytic measles virus. Viruses. 2016;8:294.
Ebert O, Shinozaki K, Huang T-G, Savontaus MJ, Garcia-Sastre A, Woo SLC. Oncolytic vesicular stomatitis virus for treatment of orthotopic hepatocellular carcinoma in immune-competent rats. Cancer Res. 2003;63:3605–11.
Balachandran S, Barber GN. Vesicular stomatitis virus (VSV) therapy of tumors. IUBMB Life. 2000;50:135–8.
Ozduman K, Wollmann G, Piepmeier JM, van den Pol AN. Systemic vesicular stomatitis virus selectively destroys multifocal glioma and metastatic carcinoma in brain. J Neurosci. 2008;28:1882–93.
Wollmann G, Rogulin V, Simon I, Rose JK, van den Pol AN. Some attenuated variants of vesicular stomatitis virus show enhanced oncolytic activity against human glioblastoma cells relative to normal brain cells. J Virol. 2010;84:15163–1573.
Zhang X, Mao G, van den Pol AN. Chikungunya-vesicular stomatitis chimeric virus targets and eliminates brain tumors. Virology. 2018;522:244–59.
Jiang B, Huang D, He W, Guo W, Yin X, Forsyth P, et al. Inhibition of glioma using a novel non-neurotoxic vesicular stomatitis virus. Neurosurg Focus. 2021;50:E9.
Allen C, Paraskevakou G, Liu C, Iankov ID, Msaouel P, Zollman P, et al. Oncolytic measles virus strains in the treatment of gliomas. Expert Opin Biol Ther. 2008;8:213–20.
Lal S, Carrera D, Phillips JJ, Weiss WA, Raffel C. An oncolytic measles virus-sensitive Group 3 medulloblastoma model in immune-competent mice. Neuro Oncol. 2018;20:1606–15.
Studebaker AW, Kreofsky CR, Pierson CR, Russell SJ, Glanis E, Raffel C. Treatment of medulloblastoma with a modified measles virus. Neuro Oncol. 2010;12:1034–42.
Allen C, Opyrchal M, Aderca I, Schroeder MA, Sarkaria JN, Domingo E, et al. Oncolytic measles virus strains have a significant antitumor activity against glioma stem cells. Gene Ther. 2013;2:444–9.
Msaouel P, Dispenzieri A, Galanis E. Clinical testing of engineered oncolytic measles virus strains in the treatment of cancer: An overview. Curr Opin Mol Ther. 2009;11:43–53.
Viral Therapy in Treating Patients with Recurrent Glioblastoma Multiforme. www.clinicaltrials.govNCT00390299 (Accessed on September 7, 2021).
Ebert O, Harbaran S, Shinozaki K, Woo SLC. Systemic therapy of experimental breast cancer metastases by mutant vesicular stomatitis virus in immune-competent mice. Cancer Gene Ther. 2005;12:350–8.
Levelle S, Samuel S, Goulet M-L, Hiscott J. Enhancing VSV oncolytic activity with an improved cytosine deaminase suicide gene strategy. Cancer Gene Ther. 2011;18:435–43.
Sugiyama T, Yoneda M, Kuraishi T, Hattori S, Inoue Y, Sato H, et al. Measles virus selectively blind to signaling lymphocyte activation molecule as a novel oncolytic virus for breast cancer treatment. Gene Ther. 2013;20:338–47.
Abdullah SA, Al-Shammari AM, Lateef SA. Attenuated measles vaccine strain have potent oncolytic activity against Iraqi patient derived breast cancer cell line. Saudi J Biol Sci. 2020;27:865–72.
Yang B, Shi J, Sun Z, Zhu D, Xu X. Attenuated measles virus overcomes radio- and chemoresistance in human breast cancer cells by inhibiting the non-homologous end joining pathway. Oncol Rep. 2020;44:2253–64.
World Health Organization Human papillomavirus vaccines: WHO position paper, May 2017. Wkly Epidemiol Rec. 2017;92:241–68.
Cantarella G, Liniger M, Zuniga A, Schiller JT, Billeter M, Naim HY, et al. Recombinant measles virus-HPV vaccine candidates for prevention of cervical carcinoma. Vaccine. 2009;27:3386–90.
Gupta G, Giannino V, Rishi N, Glueck R. Immunogenicity of next-generation HPV vaccines in non-human primates: Measles-vectored HPV vaccine versus Pichia pastoris recombinant protein vaccine. Vaccine. 2016;34:4724–31.
Brandsma JL, Shylankevich M, Su Y, Roberts A, Rose JK, Zelterman D, et al. Vesicular stomatitis virus-based therapeutic vaccination targeted to the E1, E2, E6, and E7 proteins of cottontail rabbit papillomavirus. J Virol. 2007;81:5749–58.
Liao JB, Publicover J, Rose JK, DiMaio D. Single-dose, therapeutic vaccination of mice with vesicular stomatitis virus expressing human papillomavirus type 16 E7 protein. Clin Vaccine Immunol. 2008;15:817–24.
Grossardt C, Engeland CE, Bossow S, Halama N, Zaoui K, Leber MF, et al. Granulocyte-macrophage colony-stimulating factor-armed oncolytic measles virus is an effective therapeutic cancer vaccine. Hum Gene Ther. 2013;24:644–54.
Backhaus PS, Veinalde R, Hartmann L, Dunder JE, Jeworowski LM, Albert J, et al. Immunological effects and viral gene expression determine the efficacy of oncolytic measles vaccines encoding IL-12 or IL-15 agonists. Viruses. 2019;11:914.
Wang, J, Liu, T, Chen, J. Oncolytic measles virus encoding Inreleukin-12 mediated antitumor activity and immunologic control of colon cancer in vivo and ex vivo. Cancer Biother Radiopharm. 2020. https://doi.org/10.1089/cbr.2019.3084.
Stephenson KB, Barra NG, Davies E, Ashkar AA, Lichty BD. Expressing human interleikin-15 from oncolytic vesicular stomatitis virus improves survival in a murine metastatic colon adenocarcinoma model through the enhancement of anti-tumor immunity. Cancer Gene Ther. 2012;19:238–46.
Yamaki M, Shinozaki K, Sakaguchi T, Meseck M, Ebert O, Ohdan H, et al. The potential of recombinant vesicular stomatitis virus-mediated virotherapy against metastatic colon cancer. Int J Mol Med. 2013;31:299–306.
Day GL, Bryan ML, Northrup SA, Lyles DS, Westcott MM, Stewart JH 4th. Immune effects of M51R vesicular stomatitis virus treatment of carcinomatosis from colon cancer. J Surg Res. 2020;245:127–35.
Zhao D, Chen P, Yang H, Wu Y, Zeng X, Zhao Y, et al. Live attenuated measles virus vaccine induces apoptosis and promotes tumor regression in lung cancer. Oncol Rep. 2013;29:199–204.
Boisgerault N, Guillerme JB, Pouliquen D, Mesel-Lemoine M, Achard C, Combredet C, et al. Natural oncolytic activity of live-attenuated measles virus against human lung and colorectal adenocarcinomas. Biomed Res Int. 2013;2013:387362.
Patel MR, Jacobson BA, Belgum H, Raza A, Sadiq A, Drees J, et al. Measles vaccine strains for virotherapy of non-small cell lung carcinoma. J Thorac Oncol. 2014;9:1101–10.
Patel MR, Jacobson BA, Ji Y, Drees J, Tang S, Xiong K. Vesicular stomatitis virus expressing interferon-β is oncolytic and promotes antitumor immune responses in a syngeneic murine model of non-small cell lung cancer. Oncotarget. 2015;6:33165–77.
Patel MR, Dash A, Jacobson BA, Ji Y, Baumann D, Ismail K, et al. JAK/STAT inhibition with ruxolitinib enhances oncolytic virotherapy in non-small cell lung cancer models. Cancer Gene Ther. 2019;26:411–8.
Schreiber L-M, Urbiola C, Das K, Spiesschaert B, Kimpel J, Heinemann F, et al. The lytic activity of VSV.GP treatment dominates the therapeutic effects in a syngeneic model of lung cancer. Br J Cancer. 2019;121:647–58.
Pasquinucci G. Possible effect of measles on leukaemia. Lancet. 1971;297:136.
Zygiert Z. Hodgkin’s disease: Remissions after measles. Lancet. 1971;297:593.
Lühl NC, Zimgibl F, Dorneburg C, Wei J, Dahlhaus M, Barth TFE, et al. Attenuated measles virus controls prediatric acute B-lineage lymphoblastic leukemia in NOD/SCID mice. Haematologica. 2014;99:1050–61.
Maurer S, Salih HR, Smirnow I, Lauer UM, Berchtold S. Suicide gene-armed measles vaccine virus for the treatment of AML. Int J Oncol. 2019;55:347–58.
Cesaire R, Oliere S, Sharif-Askari E, Loignon M, Lezin A, Olindo S, et al. Oncolytic activity of vesicular stomatitis virus in primary adult T-cell leukemia. Oncogene. 2006;25:349–58.
Shen W, Patnaik MM, Ruiz A, Russell SJ, Peng K-W. Immunotherapy with vesicular stomatitis virus and PD-L1 blockade enhances therapeutic outcome in murine acute myeloid leukemia. Blood. 2016;127:1449–58.
Samuel S, Tumilasci VF, Oliere S, Nguyen TL-A, Shamy A, Bell J, et al. VSV oncolysis in combination with the BCL-2 inhibitor obatoclax overcomes apoptosis resistance in chronic lymphocytic leukemia. Mol Ther. 2010;18:2094–103.
Grote D, Russell SJ, Cornu TI, Cattaneo R, Vile R, Poland GA, et al. Live attenuated measles virus induces regression of human lymphoma xenografts in immunodeficient mice. Blood. 2001;97:3746–54.
Miest TS, Frenzke M, Cattaneo R. Measles virus entry through the signaling lymphocyte activation molecule governs efficacy of mantle cell lymphoma radiovirotherapy. Mol Ther. 2013;21:2019–31.
Heinzerling L, Künzi V, Oberholzer PA, Kündig T, Naim H, Dummer R. Oncolytic measles virus in cutaneous T-cell lymphomas mounts antitumor immune responses in vivo and targets interferon-resistant tumors. Blood. 2005;106:2287–94.
Hanauer JDS, Rengst B, Kleinlützum D, Reul J, Pfeiffer A, Friedel T, et al. CD30-targeted oncolytic viruses as novel therapeutic approach against classical Hodgkin lymphoma. Oncotarget. 2018;9:12971–81.
Kaufmann JK, Bossow S, Grossardt C, Sawall S, Kupsch J, Erbs P, et al. Chemovirotherapy of malignant melanoma with a targeted and armed oncolytic measles virus. J Invest Dermatol. 2013;133:1034–42.
Allaqui F, Achard C, Panterne C, Combredet C, Labarrière N, Dréno B, et al. Modulation of the type I interferon response defines the sensitivity of human melanoma cells to oncolytic measles virus. Curr Gene Ther. 2017;16:419–28.
Ammour Y, Ryabaya O, Shchetinina Y, Prokofeva E, Gavrilova M, Khochenkov D, et al. The susceptibility of human melanoma cells to infection with the Leningrad-16 vaccine strain of measles virus. Viruses. 2020;12:173.
Galivo F, Diaz RM, Thompson WJ, Kottke T, Barber G, Melcher A, et al. Single-cycle viral gene expression, rather than progressive replication and oncolysis, is required for VSV therapy of B16 melanoma. Gene Ther. 2010;17:158–70.
Kimpel J, Urbiola C, Koske I, Tober R, Banki Z, Wollmann G. The Oncolytic virus VSV-GP is effective against malignant melanoma. Viruses. 2018;10:108.
Pol JG, Zhang L, Bridle BW, Stephenson KB, Resséquier J, Hanson S, et al. Maraba virus as a potent oncolytic vaccine vector. Mol Ther. 2014;22:420–9.
Peng K-W, TenEyck CJ, Galanis E, Kalli KR, Hartmann LC, Russell SJ. Intraperitoneal therapy of ovarian cancer using an engineered measles virus. Cancer Res. 2002;62:4656–62.
Hasegawa K, Nakamura T, Harvey M, Ikeda Y, Oberg A, Figini M, et al. The use of a tropism-modified measles virus in folate receptor-targeted virotherapy of ovarian cancer. Clin Cancer Res. 2006;12:6170–8.
Hasegawa K, Pham L, O’Connor MK, Federspiel MJ, Russel SJ, Peng K-W. Dual therapy of ovarian cancer using measles viruses expressing carcinoembryonic antigen and sodium iodide symporter. Clin Cancer Res. 2006;12:1868–75.
Galanis E, Hartmann LC, Cliby WA, Long HJ, Peethambaram PP, Barrette BA, et al. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res. 2010;70:875–82.
Lin X, Chen X, Wei Y, Zhao J, Fan L, Wen Y, et al. Efficient inhibition of intraperitoneal human ovarian cancer growth and prolonged survival by gene transfer of vesicular stomatitis virus matrix protein in nude mice. Gynecol Oncol. 2007;104:540–6.
Zhong Q, Wen YJ, Yang HS, Luo H, Fu AF, Yang F, et al. Efficient cisplatin-resistant human ovarian cancer growth and prolonged survival by gene transferred vesicular stomatitis virus matrix protein in nude mice. Ann Oncol. 2008;19:1584–91.
Long J, Yang Y, Kang T, Zhao W, Cheng H, Wu Y, et al. Ovarian cancer therapy by VSVMP gene mediated by a paclitaxel-enhanced nanoparticle. ACS Appl Mater Interfaces. 2017;9:39152–64.
Dold C, Rodriguez Urbiola C, Wollmann G, Egerer L, Muik A, Bellmann L, et al. Application of interferon modulators to overcome partial resistance to ovarian cancers to VSV-GP oncolytic viral therapy. Mol Ther Oncolytics. 2016;3:16021.
Awano M, Fuijyki T, Shoji K, Amagai Y, Murakami Y, Furukawa Y, et al. Measles virus selectively blind to signaling lymphocyte activity molecule has oncolytic efficacy against nectin-4 expressing pancreatic cells. Cancer Sci. 2016;107:1647–52.
May V, Berchtold S, Berger A, Venturelli S, Burkard M, Leischner C, et al. Chemovirotherapy for pancreatic cancer: Gemcitabine plus oncolytic measles vaccine virus. Oncol Lett. 2019;18:5534–42.
Murphy AM, Besmer DM, Moerdyk-Schauwecker M, Moestl N, Ornelles DA, Mukherjee P. Vesicular stomatitis virus as an oncolytic agent against pancreatic ductal adenocarcinoma. J Virol. 2012;86:3073–87.
Hastle E, Besmer DM, Shah NR, Murphy AM, Moredyk-Schauwecker M, Molestina C, et al. Oncolytic vesicular stomatitis virus in an immunocompetent model of MUC1-positive or MUC1-nulll pancreatic ductal adenocarcinoma. J Virol. 2013;87:10283–94.
Hastie E, Cataldi M, Moerdyk-Schauwecker MJ, Felt SA, Steuerwald N, Grdzelishvili VZ. Novel biomarkers of resistance of pancreatic cancer cells to oncolytic vesicular stomatitis virus. Oncotarget. 2016;7:61601–18.
Nagalo BM, Breton CA, Zhou Y, Arora M, Bogenberger JM, Barro O, et al. Oncolytic virus with attributes of vesicular stomatitis virus and measles virus in hepatobiliary and pancreatic cancers. Mol Ther Oncolytics. 2020;18:546–55.
Msaouel P, Iankov ID, Allen C, Morris JC, von Messling V, Cattaneo R. Engineered measles virus as a novel oncolytic therapy against prostate cancer. Prostate. 2009;69:82–91.
Liu C, Hasegawa K, Russell SJ, Sadelain M, Peng K-W. Prostate-specific membrane antigen retargeted measles virotherapy for the treatment of prostate cancer. Prostate. 2009;69:1128–41.
Son HA, Zhang L, Cuong BK, Van Tong H, Cuong LD, Hang NT, et al. Combination of vaccine-strain measles and mumps viruses enhances oncolytic activity against human solid malignancies. Cancer Investig. 2018;7:106–17.
Zhao X, Huang S, Luo H, Wan X, Gui Y, Li J, et al. Evaluation of vesicular stomatitis virus mutant as an oncolytic agent against prostate cancer. Int J Clin Exp Med. 2014;7:1204–13.
Urbiola C, Santer FR, Petersson M, van der Pluijm G, Horninger W, Erlmann P. Oncolytic activity of the rhabdovirus VSV-GP against prostate cancer. Int J Cancer. 2018;143:1786–96.
Fehl DJ, Ahmed M. Curcumin promotes the oncolytic capacity of vesicular stomatitis virus for the treatment of prostate cancers. Virus Res. 2017;228:14–23.
Author information
Authors and Affiliations
Contributions
All contributions to the manuscript, including text, tables and figures are produced by the sole author.
Corresponding author
Ethics declarations
COMPETING INTERESTS
The author declares no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lundstrom, K. Self-replicating vehicles based on negative strand RNA viruses. Cancer Gene Ther 30, 771–784 (2023). https://doi.org/10.1038/s41417-022-00436-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-022-00436-7
This article is cited by
-
Self-replicating messenger RNA based cancer immunotherapy
Cancer Gene Therapy (2023)
