
Abstract
The microdomain structures and liquid crystal (LC) orientations of ABA triblock copolymers comprising poly(styrene) (PS) A blocks and a main-chain LC polyester B block were examined via small-angle X-ray scattering and electron microscopy in the form of fibrous samples. The LC polyester blocks were segregated from the PS blocks to form a nematic LC in the lamellar microdomains of the copolymers, with the PS volume fraction (φ) ranging from 13 to 38%. As φ decreased, the nematic director was always positioned along the fiber axis, but the microdomain lamellae changed their direction from parallel to perpendicular with respect to the fiber axis and began to adopt a zigzag configuration. These lamellar orientations were attributed to the main-chain nematic LC segments lying along the microdomain interface but extending perpendicularly to the interface as the occupying interface area was reduced. The lamellar microdomains parallel to the nematic director increased the LC and PS lamellar thicknesses reversibly by 48 and 16%, respectively, at the same time as the isotropization of the nematic LC. Such changes in lamellar thickness suggest that both the PS and LC segments were elongated along the direction of the LC orientation.
Similar content being viewed by others


Introduction
Liquid crystalline block copolymers (LCBCPs) consisting of liquid crystalline (LC) segments have attracted considerable attention because the self-assembled structures of the LC segment are easily oriented using an external field. A magnetic field can align amorphous microcylinders, as well as the LC director of the matrix along the field direction. The microdomain orientation is induced by the LC orientation via mesogenic units anchoring parallel to the microdomain interface. Such an LC anchoring is readily explained for the side-chain type of LC polymer segments typically adopted in LCBCPs. The mesogens arrange their long axis parallel to the interface because they are connected at the end to the segment’s backbone that stretches perpendicularly to the interface via the most extended spacers.
We previously reported on LCBCPs using the main-chain-type LC polymers as an LC segment [1,2,3,4,5,6]. The LCBCPs were ABA triblock copolymers comprising an LC polyester as the B block connected at both ends to poly(methacrylate) (PMA) A blocks. Although the LC polyesters had molecular weight distribution with a polydispersity index as large as 2, the BCP with the B block of a smectic (Sm) LC BB-5(3-Me) polyester formed a well-defined lamellar microdomain structure to display scattering maxima up to the seventh order in the small-angle X-ray scattering (SAXS) pattern. Such a lamellar microdomain was formed at a larger composition range of the PMA volume fraction between 19 and 50%. The LC lamellar microdomain consisted of an Sm LC with the LC director perpendicular to the lamellar interface [1, 2, 4]. Such an LC orientation was reasonable for main-chain LC segments incorporating mesogens within the backbone and agreed with the tendency of BCP segments extended perpendicularly to the microdomain interface. However, LC segments with contour lengths longer than the lamellar thickness must be folded several times to be accommodated in the lamellar microdomains. The LC segment increased the number of folds to augment the occupying interfacial area because the PMA segment increased the molecular weight (dimensions). When the stem length of the folded LC segment was comparable to the persistence length, the LC segment could not increase the number of foldings. Therefore, the segment was inclined with respect to the lamellae to increase the interface area [3].
An ABA BCP composed of the B block of a main-chain nematic HBA10THQ LC polyester connected to the A segments of poly(styrene) (PS) formed a lamellar structure at a PS volume fraction of 0.45 [6]. Although the LC segment was segregated from PS to form lamellae, the nematic director was parallel to the lamellae, indicating that the LC segment was elongated along the lamellar boundary. This LC segment orientation was not expected from the tendency of the BCP segments to elongate away from the microdomain interface but could be a feature of a flexible main-chain LC polymer with longer spacers. HBA10THQ has a longer spacer of decane methylene connecting two consecutive mesogens in the backbone than BB-5(3-Me), which has a spacer backbone that consists of five carbon atoms.
In this study, we synthesized a series of ABA triblock T5PS-φ copolymers using a main-chain nematic LC HBA5THQ polyester with a pentamethylene spacer (Scheme 1). HBA5THQ with a number average molecular weight (Mn) of 22 k was connected to the PS segments, with Mn ranging from 3 k to 12 k. The LCBCPs at the PS volume fraction (φ) ranging from 13 to 38% formed lamellar microdomains. The LCBCP with φ > 34% formed flat lamellae, with the nematic director parallel to the lamellae. As φ decreased, the flat lamellae became zigzag at φ = 29%, and the zigzag lamellae ran perpendicularly to the nematic director at φ = 13%.

Chemical structure of T5PS-φ
Experimental
Materials
The central HBA5THQ block was prepared by melt transesterification of diphenyl 4,4′-(pentane-1,5-diylbis(oxy))dibenzoate and an excess of tert-butylhydroquinone using a sodium carbonate catalyst at 200–260 °C [7]. The α,ω-di-2-bromoisobutyryl-terminated HBA5THQ was prepared by reacting the hydroxyl-terminated HBA5THQ with 2-bromo-2-methylpropionyl bromide. The T5PS-φ copolymers were prepared by the atom transfer radical polymerization of styrene at 110 °C using anisole and α,ω-di-2-bromoisobutyryl-terminated HBA5THQ as a solvent and a macroinitiator, respectively [6]. The experimental details are provided in the Supplementary Information. The degree of polymerization (DP) and the number average molecular weights (Mn,LC and Mn,am) of the central LCP and amorphous groups were estimated by 1H-NMR spectra. The PS volume fraction (φ) was estimated on the basis of the densities (1.19 g cm−3 for HBA5THQ; 1.05 g cm−3 for PS). The polydispersity indices (PDIs) of the copolymers were obtained by size exclusion chromatography (SEC) (Table 1).
Methods
1H-NMR spectra were recorded on a JEOL AL-400 spectrometer at 20 °C. Chemical shifts were reported in units of ppm downfield from an internal reference SiMe4. SEC was performed using a JASCO system equipped with PS gel columns with CHCl3 as the eluent after calibration with standard PS. Differential scanning calorimetry (DSC) was performed using a Perkin-Elmer Pyris 1 DSC calorimeter at a scanning rate of 10 °C min−1 in a flow of dry nitrogen. Wide-angle X-ray diffraction (WAXD) patterns were obtained using a Bulker D8 DISCOVER equipped with a Vantec-500 detector and Cu Kα radiation. SAXS patterns were recorded using a Bruker AXS Nano-STAR-U instrument. Synchrotron radiation (SR) SAXS measurements were performed at the BL-6A beam line in Photon Factory (Tsukuba, Japan) equipped with a Dectris PILATUS300K-W detector with a camera length of ~2 m. The X-ray wavelength was 0.15 nm. The scattering intensity was corrected by the transmission and subtraction of background scattering and plotted against the scattering vector q = (4π sin θ)/λ. Transmission electron microscopy (TEM) was performed using a Hitachi H-7650 Zero A electron microscope operating at 100 kV in the bright-field mode. Samples were exposed to RuO4 vapors at ambient temperature to selectively stain the LC domains and then microtomed into ultrathin sections.
Results
The LCP segments of T5PS-φ and the precursor hydroxyl-terminated HBA5THQ exhibited phase transitions similar to an HBA5THQ homopolymer [7]. The hydroxyl-terminated HBA5THQ exhibited the glass transition and the isotropization of the nematic LC at 91 and 183 °C, respectively, as observed in the heating DSC thermogram. The heating DSC thermograms of the BCPs included an endothermic peak and a step in the heat capacity that were ascribed to the isotropization of the nematic LC and the glass transition, respectively. The isotropization temperature (Ti) and the glass transition temperature (Tg) ranged from 162 to 182 °C and from 81 to 90 °C, respectively (Table 1). Thus, although the nematic LC segments were segregated from the PS segments, the copolymers exhibited a single glass transition because the nematic LC and PS segments were similar in Tg. The isotropization enthalpy (ΔHi) of the LC block was 1 kJ mol−1, which is half that of the hydroxyl-terminated HBA5THQ.
The LC segment formed a nematic LC as did the HBA5THQ homopolyester [7]. Figure 1 shows a typical two-dimensional (2D) WAXD pattern for fibrous T5PS-38. All the fibrous samples were spun from the isotropic melt at 190 °C, subsequently annealed for 4 h at a temperature of 20 °C lower than Ti, and cooled to 25 °C. The pattern includes a broad reflection with a spacing of 0.45 nm concentrated on the equator, which is ascribed to the mesogens lying parallel to the fiber axis and indicates that the LC segment formed a nematic phase with a parallel director to the fiber axis.

WAXD pattern of fibrous T5PS-38. The fiber axis is vertical
All the fibrous copolymers displayed similar WAXD patterns, indicating that the nematic director was aligned along the fiber axis. However, the corresponding SAXS patterns exhibited different azimuthal directions of the maxima among the copolymers. All the copolymers except T5PS-13 displayed scattering maxima at scattering vectors (q) with integer ratios. T5PS-13 exhibited scattering maxima up to the second order only for a non-oriented sample (Figure S2). Thus, the T5PS-φ copolymers formed lamellar microdomains. However, the normal direction with respect to the nematic director orientation was dependent on φ. T5PS-38 and T5PS-34 displayed SAXS maxima concentrated on the equator perpendicular to the fiber axis (Fig. 2a, b). The SAXS pattern did not change as φ was decreased to 34%. However, T5PS-29 exhibited scattering maxima azimuthally extended above and below the equator (Fig. 2c), suggesting that the lamellae adopted a zigzag conformation. The corresponding TEM image contained zigzag stripes running along the fiber axis (Fig. 2e). In addition, when φ was reduced to 13%, the scattering maxima spot appeared at a larger azimuth from the equator (Fig. 2d). Such a SAXS maximum in the quadrant could be ascribed to one of the following two different types of lamella. One type was the lamellae parallel to the fiber axis making a sharper zigzag than the lamellae in T5PS-29. The other was the lamellae running in a zigzag perpendicular to the fiber. The latter was found in the TEM images of a fibrous T5PS-13 sample. The typical image shown in Fig. 2f includes stripes running in zigzags perpendicular to the fiber axis. The zigzag vertex angle was 90°, equal to the supplementary angle of 90° at which the scattering maxima were split. Therefore, the lamellae adopted the zigzag configuration running perpendicularly to the fiber axis. These results demonstrate that T5PS-φ changed the lamellar orientation with respect to the LC director orientation.

SAXS patterns of fibrous T5PS-38 (a), T5PS-34 (b), T5PS-29 (c), and T5PS-13 (d) samples, and the corresponding TEM images measured for T5PS-29 (e) and T5PS-13 (f). The fiber axis is vertical
The thicknesses of the LC and PS lamellae (dLC and dam, respectively) and the standard deviations of each lamellar thickness (σLC and σam), as well as the lamellar spacing (d0) were estimated by numerically fitting the SAXS intensity using the paracrystal theory [8]. The intensity profiles shown in Fig. 3 were obtained by averaging the intensity of the 2D pattern over the azimuthal sectors of 10° on each side of the direction in which the scattering maxima were aligned. The parameter fitting values for the profiles are listed in Table 2. Here, dam was assumed to be smaller than dLC because φ is less than 50%. The characteristic of the copolymers was that d0 decreased as the molecular weight of the copolymer increased.

SAXS intensity profiles of T5PS-φ against the scattering vector q (dots). The solid curves indicate the calculated intensity based on the paracrystal theory. Arrows indicate the positions of the scattering maxima
Discussion
The LC blocks in all the fibrous copolymers invariably positioned the nematic director along the fiber axis. However, the lamellar microdomain arrangement was dependent on φ. Here, we discuss how an LC segment was accommodated in the lamellar microdomain for the three typical T5PS-φ copolymers: T5PS-38, T5PS-29, and T5PS-13. The length of the LC segment contour was estimated as 102 nm on the basis of the repeating unit length (2.23 nm) and the DP (m = 46).
T5PS-38
T5PS-38 formed lamellae parallel to the fiber axis so that the nematic director was parallel to the lamellar boundary. In the LC lamellae, the LC segments should be elongated along the nematic director. Figure 4 depicts a plausible conformation of the LC segment accommodated in an LC block lamella. An LC segment with the contour length of 102 nm primarily stretches along the nematic direction but folds to meander through a 13-nm-thick LC lamella and bridges the PS lamellae. In the amorphous block, the PS segment could extend along the boundary because both segments must occupy the same interface area to form a flat lamellar boundary.

Schematic of lamellar microdomains in T5PS-38
This lamellar structure was affected by the nematic order in the LC block. The lamellar structure of the copolymer with the LC segment in the isotropic liquid phase had different thicknesses and different spacings. The dLC and dam values were estimated as 20.0 and 10.8 nm, respectively, on the basis of the SAXS profile measured for the non-oriented sample annealed at 200 °C for 10 min and then quenched (Figure S3). The dLC and dam measured for the bulk copolymer with the LC segment in the nematic phase were 13.5 and 9.3 nm, respectively (Table S1); these lengths are comparable to that of the fibrous sample (Table 2). Thus, the dLC and dam in the copolymer with the LC segment in the isotropic phase are greater than those estimated for the same copolymer with the LC segment in the nematic phase. The increase in dLC was 48%, which is three times that of dam (16%), suggesting that the nematic order preferentially elongated the LC segment over the PS segment.
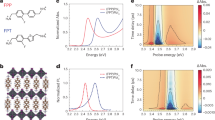
Such increases in dLC and dam occurred reversibly at the same time as the isotropization of the LC segment. The SR-SAXS profiles of T5PS-38 were measured during heating and cooling at a rate of 2 °C min−1 as they crossed the Ti of the LC segment. The profiles include the first- and second-order scattering maxima (Fig. 5a), indicating that the lamellar morphology was conserved during the heating/cooling cycle. The first-order maximum at q = 0.3 nm−1 shifted toward a smaller q = 2.5 nm−1 as the temperature increased from 180 to 200 °C and then returned to its original position with a subsequent temperature decrease. The estimated d0 from the first-order scattering maximum was plotted against the temperature (Fig. 5b). The lamellar spacing increased the isotropization of the LC segment reversibly from 24.2 to 28.5 nm. A similar reversible increase in d0 was observed for homologous copolymers with different LC spacer lengths [6].

a Sets of SR-SAXS profiles of the non-oriented T5PS-38 copolymer recorded during heating and cooling at a rate of 2.0 °C min−1. Each profile was obtained by irradiating the sample for 119 s. The thick lines indicate the profiles at an interval of 20 °C. b Lamellar spacing determined by the SAXS profiles against temperature. The red and blue circles indicate the heating and cooling process, respectively
T5PS-29
T5PS-29 formed elongated PS segments along the lamellar boundary and aligned the interfacial area with the LC segments to form the lamellar microdomains. Such an elongated PS conformation could be explained by the logarithmic plot of dam against Mn,am (Fig. 6). For T5PS-φ with φ ≥ 29%, dam increased with Mn,am according to the relationship dam∼Mn,am0.42. The index of 0.42 was considerably smaller than the normally estimated index for amorphous BCPs [9] (~M2/3) and that of Gaussian chains, suggesting that PS segments increased their dimensions with increasing Mn,am preferentially along the lamellar boundary. Although the PS segments increased the dimensions to enlarge dam, dLC decreased more markedly to decrease d0 irrespective of the increase in the molecular weight of the copolymer. T5PS-13 had greater d0 than that expected from extrapolation of the relationship between the three T5PS-φ copolymers with φ ≥ 29%.

Lamella spacing d0 (closed circles) and thickness of LC and PS lamellae (dLC and dam) (open circles and closed triangles, respectively) as a function of Mn,am in the fibrous samples. The slopes of the lines are −0.19, −0.51, and 0.42 for d0, dLC, and dam, respectively
T5PS-29 differed from T5PS-38 and T5PS-34 in lamellar microdomain morphology. The lamellae meandered with tilted boundaries with respect to the nematic director to balance the interface areas occupied by each segment. As Mn,am decreased, the PS segment could occupy a smaller interfacial area, whereas the LC segment with a constant Mn,LC tended to occupy the same interfacial area. Such a mismatch of the occupying interfacial areas between two incompatible segments might be resolved by tilting the interface with respect to the segment direction to form lamellar microdomains with segments segregated from one another.
T5PS-13
T5PS-13 was distinct from other T5PS-φ copolymers in the lamellar stacking direction with respect to the fiber axis. The lamellae were stacked along the fiber axis with a tilting angle of ±40° with respect to the stacking direction and assumed a zigzag form. Thus, fibrous T5PS-φ changed the lamellar stacking direction by 90° as Mn,am decreased, whereas the nematic director orientation remained parallel to the fiber axis, as confirmed by the WAXD pattern (Figure S4).
Such a change in the lamellar stacking direction in fibrous T5PS-φ could be attributed to the preferred LC orientation at the lamellar microdomain interface. The LC mesogen orientation at the microdomain interface could determine the LC orientation in the microdomain. This phenomenon is similar to the “LC anchoring effect,” whereby the LC orientation in an LC display cell is determined by the LC anchoring onto the cell substrate surface. By modifying the surface with the coating, the LC orientation direction in a cell could be controlled to be parallel, perpendicular, or inclined to the substrate surface. In the T5PS-φ copolymers, the LC anchoring could be associated with the relationship of the interface area occupied by the PS segment (Si,PS) to the lateral area of the LC segment repeating unit (Sl,LC). The value of Sl,LC was estimated as 1.0 nm2, which is equal to the product of the unit length (2.23 nm) and the average lateral distance between the chains (0.45 nm). The value of Si,PS was estimated using the relationship Si,PS = Mn,am/(ρam NA dam), where ρam is the density of PS (1.05 g cm−3) and NA is Avogadro’s number. The Si,PS values in T5PS-38, T5PS-34, T5PS-29, and T5PS-13 were calculated as 2.2, 2.0, 1.8, and 0.86 nm2, respectively. Here, dam of T5PS-13 was assumed to be equal to d0 × φ/100. The LC anchoring at the lamellar interface could be classified by the magnitude relationship between Si,PS and Sl,LC. In the case of Si,PS > 2Sl,LC, the mesogens were positioned along the interface, as observed for T5PS-38 and T5PS-34. In contrast, in the case of Si,PS < Sl,LC, the mesogens were supposed to stand up, although the mesogens in T5PS-13 were inclined toward the normal lamellae. The lamellae might need to incline toward the interface to compensate for Si,PS, which was smaller than Sl,LC. The lamellar morphologies observed for these four copolymers suggest that a flat lamellar interface might be formed only when Si,PS ≥ 2Sl,LC. In other cases, the lamellae would incline toward the interface to ensure that the interfacial areas occupied by the PS segment were balanced with 2Sl,LC to form the lamellae stacking perpendicularly or parallel to the LC director. Therefore, the lamellae in T5PS-29 meandered parallel to the LC director, and those in T5PS-13 adopted a zigzag conformation perpendicular to the LC director. Lamellae lying perpendicular to the nematic director may not form flat boundaries. The LC anchoring transition on the BCP microdomain interface might resemble that predicted theoretically for the nematic solvent-swollen main-chain LC polymer brushes in increasing the grafting density [10, 11].
Conclusion
The microdomain structures of T5PS-φ copolymers consisting of nematic LC HBA5THQ and PS segments were determined by examining the fibrous samples using SAXS and TEM. In all four T5PS-φ copolymers with φ ranging between 13 and 38%, the LC segment was segregated from the PS segment to form lamellar microdomains and a nematic LC with the orientation direction parallel to the fiber axis. The microdomain lamellae were stacked in the direction perpendicular to the LC orientation direction in T5PS-φ with φ ≥ 29% but parallel to the LC orientation direction in T5PS-13. The lamellae in T5PS-29 and T5PS-13 adopted a wavy and zigzag conformation, respectively.
The variation in lamellar orientation was attributed to the transition in LC anchoring at the lamellar microdomain boundary. The nematic LC segment characteristically lay along the lamellar boundary by occupying an interfacial area larger than 2Sl,LC. Otherwise, the segment incorporating mesogens in the backbone was elongated to occupy a smaller interfacial area. Because the interfacial area increased with the dimensions (or Mn,am) of the PS segment bonded to the LC segment, the nematic LC orientation changed the director orientation from parallel to perpendicular with respect to the lamellar boundary as Mn,am decreased. The lamellae might adopt a wavy or zigzag conformation to match the interfacial areas between the segments.
The LC and PS segments were elongated along the nematic directors. For copolymers forming lamellae parallel to the nematic director, dam obeyed a power function of Mn,am0.42 with an index smaller than that of the Gaussian chains. The LC and PS lamellar thicknesses increased reversibly by 48 and 16% simultaneously with the isotropization of the LC segment.
The main-chain nematic LC segments in the BCPs tended to lie along the microdomain interface, whereas the amorphous segments tended to elongate away from the interface. Such a tendency in the segment chain orientation is characteristic of main-chain nematic LC segments and different from that of main-chain smectic LC segments. The nematic LC segment backbone could be elongated perpendicularly to the microdomain interface if the counter segment connected to the LC segment occupies an interfacial area comparable to Sl,LC. The LC anchoring transition found in this study might enlarge the design of LC elastomers in cross-linked BCPs [12].
References
Ishige R, Ishii T, Tokita M, Koga M, Kang S, Watanabe J. Well-ordered lamellar microphase-separated morphology of an ABA triblock copolymer containing a main-chain liquid crystalline polyester as the middle segment. Macromolecules. 2011;44:4586–8.
Koga M, Ishige R, Sato K, Ishii T, Kang S, Sakajiri K, et al. Well-ordered lamellar microphase-separated morphology of an ABA triblock copolymer containing a main-chain liquid crystalline polyester as the middle segment 2: influence of amorphous segment molecular weight. Macromolecules. 2012;45:9383–90.
Koga M, Abe K, Sato K, Koki K, Kang K, Sakajiri K, et al. Self-assembly of flexible-semiflexible-flexible triblock copolymers. Macromolecules. 2014;47:4438–44.
Koga M, Sato K, Kang S, Sakajiri K, Watanabe J, Tokita M. Influence of smectic liquid crystallinity on lamellar microdomain structure in a main-chain liquid crystal block copolymer fiber. Macromol Chem Phys. 2013;214:2295–2300.
Koga M, Sato K, Kang S, Tokita M. Microphase-separated morphology and liquid crystal orientation in block copolymers comprising a main-chain liquid crystalline central segment connected to side-chain liquid crystalline segments at both ends. Macromol Chem Phys. 2018;219:1–8.
Sato K, Koga M, Kang S, Sakajiri K, Watanabe J, Tokita M. Lamellar morphology of an ABA triblock copolymer with a main-chain nematic polyester central block. Macromol Chem Phys. 2013;214:1089–93.
Tokita M, Kato K, Ishige R, Okuda S, Kawauchi S, Okoshi K, et al. Unusual chain configuration of main-chain liquid crystal polyesters having Y-shaped mesogens in nematic phase. Polymer. 2011;52:5830–5.
Roe, R-J. Methods of X-ray and Neutron Scattering in Polymer Science. (Oxford University Press, Oxford, 2000).
Todo A, Hiroyukiuno, Miyoshi K, Hashimoto T, Hiromichikawai. Domain‐boundary structure of styrene‐isoprene block copolymer films cast from solutions. III. Preliminary results on spherical microdomains. Polym Eng Sci. 1977;17:587–97.
Halperin A, Williams DRM. Liquid crystalline brushes: an anchoring transition. Europhys Lett. 1993;21:575–80.
Halperin A, Williams DRM. Liquid crystalline polymers in nematic solvents: interfacial behaviour and active anchoring. J Phys Condens Matter. 1994;6:A297–A300.
Abe K, Koga M, Wakabayashi T, Kang S, Sakajiri K, Watanabe J, et al. Thermally reversible distortion observed for triblock copolymers comprising main-chain liquid crystal polyesters attached to photo-cross-linked cinnamate segments at both ends. Macromolecules. 2015;48:8354–60.
Acknowledgements
This study was supported by the Strategic Promotion of Innovative Research and Development of the Japan Science and Technology Agency, JST. The SR-SAXS measurements were performed under approval of the Photon Factory Program Advisory Committee (No. 2016G612). TEM studies were performed by Mr. Jun Koki (Technical Department, Tokyo Institute of Technology), whose assistance is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Koga, M., Wakabayashi, T., Kang, S. et al. Transition of liquid crystal anchoring at the microdomain interface observed for main-chain nematic polyester segments of block copolymers. Polym J 51, 295–302 (2019). https://doi.org/10.1038/s41428-018-0131-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41428-018-0131-x
