
Abstract
We previously reported that functionalized phenyl- and vinyl-silsesquioxanes (SQs) and [RSiO1.5]8,10,12 (R = Ph or vinyl) exhibited redshifted absorption and emission, suggesting 3-D conjugation via a cage-centered lowest unoccupied molecular orbital (LUMO). The functionalized [PhSiO1.5]7(OSiMe3)3 with a missing corner and edge-opened, end-capped [PhSiO1.5]8(OSiMe2)2 (double decker, DD) analogs also exhibit emission redshifts, indicating 3-D conjugation. DD [PhSiO1.5]8(OSiMevinyl)2 and R-Ar-Br copolymers exhibit polymerization (DP)-dependent emission λmax and integer charge transfer (ICT) to 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TNCQ). The terpolymer-averaged redshifts all suggest conjugation with two (O-Si-O) endcaps, possibly via a cage-centered LUMO. In assessing conjugation limits, it was anticipated that copolymers of the ladder (LL) SQ, (vinylMeSiO2)[PhSiO1.5]4(O2SiMevinyl), with Br-Ar-Br and without a cage would eliminate LUMO formation and a redshift. The λmax values observed were greater for analogous copolymers, which requires a different explanation. Here, we assess the photophysical behavior of copolymers closer to polysiloxanes, namely, the expanded cage (MeVinylSiO)2[PhSiO1.5]8(OSiMeVinyl)2SQs. Copolymers with Br-Ar-Br exhibit redshifted absorption and emission, which supports conjugation via Si-O-Si bonds rather than cage-centered LUMOs, contrary to traditional views of Si-O-Si copolymers. One- and two-photon photophysical probes showed that XDD copolymers exhibit multiple fluorescence-emitting excited states, in violation of Kasha’s rule stating that emission should occur only from the lowest excited state. Finally, new modeling studies suggested that conjugation derives from Si-O-Si bond dπ-pπ interactions, an unexpected result for polysiloxanes that supports two forms of conjugation.
Similar content being viewed by others


Introduction
Silicon sits just below carbon in the periodic table and is expected to exhibit similar properties. Thus, silicon is known to form multiple bonds with other elements but typically in a constrained environment [1,2,3]. Unlike carbon, silicon has long been known to expand its coordination sphere by forming five and six bonds, typically with electron-donating moieties, including halides, oxygen and nitrogen [4,5,6,7]. The bonding in these systems is apparently a combination of covalent and partially ionic bonds, but is not a consequence of hypervalence [8]. Interactions of the Si σ* orbitals have also been observed in such molecules [9].
In keeping with this latter observation, silicon was also found to participate in π conjugation in siloles via σ*−π* bonding [10], which differs from the conjugation in carbon systems, which typically involves pπ-pπ interactions. Polysilanes still offer a form of conjugation not seen with all carbon systems, σ−σ* conjugation [10,11,12,13]. In the present work, we discuss other bonding interactions for silsesquioxanes and polysiloxanes that lead to apparent conjugation via mechanisms not found previously.
Polysiloxanes, silsesquioxanes in particular, have been studied extensively because they have multiple properties, including high temperature stability, biocompatibility, hydrophobicity, transparency, and insulating capacity. Widespread use has led to multiple reviews and one book [14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31]. The first modeling studies of the basic silsesquioxane (SQ) unit [HSiO1.5]8 identified the formation of a cage-centered LUMO [32, 33]. Stabilization of a cage-encapsulated F- was attributed to a cage-centered LUMO, which provided the first experimental support [34, 35]. We reported that spherical, cage-centered magnetic fields form in SQs exposed to intense laser light, providing additional experimental support for the existence of cage-centered LUMOs [36]. Support for conjugation via a cage-centered LUMO was also demonstrated with two-photon absorption (2PA) spectroscopy, wherein the absorption cross sections for the series 4-Me2NStilbeneSi(OSiMe3)3 (corner), [4-Me2NStilbeneSi(O)(OSiMe3)]4 (half-cage), and [4-Me2NStilbeneSiO1.5]8 (cage) were 12:8:26 per functional group, supporting 3-D conjugation, also indicated by modeling studies [37].
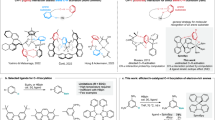
We previously reported that phenyl- and vinyl-silsesquioxanes (SQs), [RSiO1.5]8,10,12 (R = Ph or vinyl) functionalized with three or more conjugated moieties, exhibit redshifted absorption and emission features that suggest 3-D conjugation likely via a cage-centered LUMO. In an effort to find limiting structures wherein cage LUMOs did not form and evidence that conjugation is absent, we synthesized [PhSiO1.5]7(OSiMe3)3 without a corner or the edge-opened, end-capped [PhSiO1.5]8(OSiMe2)2 (double decker, DD) functionalized analogs that likewise present a redshifted spectrum, again indicating 3-D conjugation and a cage-centered LUMO (Fig. 1) [36, 38, 39].

Selected SQ structural formats
Although the main absorption onsets for almost all of the functionalized SQ compounds synthesized in these studies show very small redshifts, these redshifts may be accompanied by redshifted lower-intensity features, thus deviating from expectations based on well-studied conjugated organic polymers.
In subsequent generations, we copolymerized MeVinylSi(O-)2 end-capped DD and LL systems with multiple aromatic agents, as shown in Fig. 2a, b [40, 41] to test the conjugation limits by using divinylbenzene tethers, as earlier studies of phenyl T10 and T12 cage copolymers showed that these copolymers were conjugated [42].

Modified SQ systems designed to identify limits where conjugation does not occur
In particular, we assumed that the absence of cages in the LL SQ copolymers would coincidentally prevent formation of a LUMO and conjugation. To our surprise, both the DD and LL SQ copolymers show conjugation even with vinyldisiloxane end caps and for LL systems without cages [41]. In addition to providing photophysical evidence for conjugation, the DD and LL polymers exhibited integer electron transfer to F4TCNQ, forming F4TCNQ-, which also provides proof of conjugation. Likewise, alternating terpolymers show redshifted emissions that are the averages of the two homocopolymers [43, 44].
In the present work, we explored further deviations from the initial structures by searching for limiting siloxane/SQ oligomers/polymers in which conjugation is absent. To this end, we expanded the siloxane DD end caps (Vy4XDD, Scheme 1 trans-E isomer shown) to emphasize/introduce additional siloxane units and increase the similarity to polysiloxanes [45, 46]. We again found evidence of conjugation in the resulting copolymers. Our results, supported by 2PA measurements and modeling, pointed to a second form of conjugation via Si–O–Si bonds, as detailed below.

Vinylmethylsiloxane capped expanded cage (Vy4XDD) model compound reaction (1) and copolymerization reaction (2). Ar: selected aromatic moieties
Synthesis and characterization
Catalytic cross coupling of 1:1 molar ratios of Vy4XDD SQs and dibromoaryl (Br-Ar-Br) were performed with previously described protocols. Model compounds with the simplest conjugated structures were synthesized for comparison via Heck catalytic cross coupling of Vy4XDD SQs with monobromoaromatic (Br-Ar) molecules (Scheme 1). With four vinyl groups, cross coupling was anticipated to give polymers consisting of both linear and branched segments, perhaps limiting the potential for conjugation. As shown by the MALDI, GPC, FTIR, TGA, DTA and 1H NMR data presented in Figs. S1–S19, the anticipated mixed structures were produced (see Figs. S7 and S8). GPC and TGA data are presented in Table S1.
However, when characterizing the purified mixtures, we again observed evidence of conjugation even in the isolated solids (Fig. S20), which revealed redshifted absorption and emission compared with those of model compounds with four functional groups (Figs. 3 and S21–23), the data for which are collated in Table 1.

a Normalized absorption and emission of XDDcoStil and XDD(Stil)4. b Normalized absorption and emission for XDDcoTh with the model compound XDD(Th)4 (excitation, 265 nm/DCM)
Both the XDDcoStil and XDDcoTh absorption and emission spectra exhibit significant emission redshifts compared with those of models. Figure 3 also reveals that for XDDcoTh, where the Stokes shift was the largest, the corresponding absorption spectra show, in addition to the main peak, a relatively broad and weaker redshifted shoulder. While the main band is most likely closely related to the S0 → S1 transition of the corresponding chromophore, the mechanisms for formation of the shoulder are much less obvious.
To explore conjugation of the XDD copolymers, column chromatography was used to separate fractions of the XDDcoStil oligomer mixture and assess the effects of chain length on the photophysical properties. The original mixture ranged from monomers up to hexamers, as determined by MALDI, but materials with higher DPs may also be present but not apparent because they do not ionize efficiently, as shown in Fig. S24.
Figure 4a, b shows optical images of the column separation process and the individual samples collected as elution continued. The absorption and emission data, along with the MALDI data, are shown in Table 2. Samples marked with large numbers contain oligomers with higher DPs. The colors produced with 365 nm irradiation varied from purple to blue to greenish blue within the column and the isolated samples. In the isolated samples, the absorption λmax values were redshifted from 276 (monomer) to 356 nm (oligomer), as shown in Fig. 4c. Similarly, the emission λmax values were redshifted from 385 to ~470 nm, as shown in Fig. 4d.

a XDDcoStil excited at 365 nm showing changes in emission vs. the position on the column as a function of DP (see below). b Selected fractions from column chromatography. (the color changes from purple to greenish blue from right to left). c Absorption (solid lines) and (d) emission (dashed lines) spectra of selected samples of XDDcoStil in DCM after column chromatographic separation
In comparing the emission spectra with the associated MALDI data, new emission peaks appearing at λmax = 410, 439, and 475 nm are assigned to samples in which the dominant components are dimers, followed by trimers and tetramers to hexamers. These results offer additional proof of extended conjugation that is DP-dependent, a typical result for conjugated polymeric systems.
The series of relatively weak absorption peaks at 260–280 nm in Fig. 4c likely derive from vibronic progression of the attached phenyl groups. Interestingly, however, the amplitudes of these features when scaled relative to the main 355 nm absorption peak appear to decrease with increasing order of chromatographic fraction. The data reveal that the dimers have the highest quantum yields. When the proportion of oligomeric DPs increases, the quantum yields decrease. Sample 15 exhibited a quantum yield of 48%, which might have been caused by emission from low concentrations of dimers.
Coincidentally, most of the copolymers react with F4TCNQ via integer electron transfer, as indicated by both changes in the UV‒Vis spectrum (Figs. 5, S31) and νC≡N frequency shifts (Fig. S32 and Table S2), in accordance with our previous studies [41].

CT experiments with mixed XDD copolymers (a) without and (b) with F4TCNQ in DCM. XDDcoPh, XDDcoBiph, XDDcoTerph, XDDcoStil, XDDcoTh
2D absorption-emission spectra
Kasha’s rule states that on photoexcitation, molecules/polymers in the excited state relax to the lowest-energy excited state before relaxing to the ground state with emission of a low energy photon [47, 48]. Most of the polymers synthesized here seemed to violate this rule, as exemplified by the following data.
Given that the thiophene copolymers exhibit the most significant redshifts, we explored their photophysics in detail by generating 2D absorption-emission spectral maps for both one-photon absorption (1PA) and two-photon absorption (2PA). Figure 6a, b shows the 2PA 2D spectral map for XDDcoTh in ACN, in which the horizontal axis shows the emission wavelength (λem = 230–700 nm) and the vertical axis shows the excitation wavelength (λex = 200–550 nm). The diagonal straight lines are spectrometer artifacts. The panel on the left (right) shows the emission intensity plotted on a linear (logarithmic) scale. One can identify two distinct “origins” at which the emission wavelengths approach the excitation wavelength, λem ≈ λex, at ~420 and ~460 nm. A relatively weaker feature is observed at ~360 nm.

2D emission spectrum of XDDcoTh (ACN) with λem = 230–700 nm (horizontal axis) plotted vs. excitation wavelength, λex = 200–550 nm (vertical axis); (a) the emission intensity is shown on a linear scale and (b) on a logarithmic scale. The vertical and horizontal dashed lines indicate the locations of the three origins at 360, 420 and 460 nm. The diagonal lines are spectrometer artifacts, λem = λex, and λem = 2λex. c Log-scale 2D emission spectra of XDDcoTh in DMSO obtained by 1-photon (main panel) and 2-photon excitation (insert). The right vertical axis shows the 2-photon excitation range, λex (2PA) = 680–1000 nm. The vertical and horizontal dashed lines indicate the origins at 430 and 470 nm, respectively. The diagonal dashed line shows that λem = λex. DMSO was used to increase the solubility. d Summary of the characteristic features of the absorption and emission spectra of XDDcoTh in ACN. Blue curves—normalized absorption spectrum; Red curve –normalized emission spectrum at λex = 375 nm. The vertical bars show the frequencies of the origins observed in the 1-photon 2D excitation emission
In comparing the XDDcoTh 2D spectrum to the corresponding linear absorption profile (Fig. 3b), the weaker emission at 360–420 nm is likely due to the same electronic transition that gives rise to the main vibronic absorption band with a maximum at 345 nm, while the 420 and 460 nm origins are likely due to different electronic transitions located at lower energies. Although reliable assignments of the 420 and 460 nm origins are complicated because of substantial spectral broadening and overlap between adjacent bands, it is likely that both are related to the excited states responsible for the redshifted absorption shoulder.
It should be noted that similar absorption and emission features were observed previously, though to varying degrees, with related polymers and some model compounds [40]. The purities of the samples were determined using NMR, MALDI, FTIR, and GPC coupled with TLC. Furthermore, the fact that the observed spectral features are consistent from sample to sample and that we used different solvents provides additional assurance for our purification and analytical characterization procedures. Therefore, we did not consider sample contamination, e.g., contamination with a highly fluorescent impurity (or impurities), and interpret the above observations as evidence of behavior that does not conform to Kasha’s rule.
Figure 6c shows the 2PA-excited 2D emission intensity map for XDDcoTh in DMSO. The 2-photon excitation wavelengths are shown on the right vertical axis and span 680–1000 nm, while the corresponding fluorescence appears at 400–660 nm. The 2-photon spectral map overlay the conventional (1-photon) 2D spectrum measured in the same solvent but with a slightly broader range of wavelengths.
While 2-photon and 1-photon excitation profiles both exhibit absolute maxima near λex (1PA) = 360–370 nm [λex (2PA) = 720–740 nm], the corresponding absolute maximum emission intensity of the 2-photon spectrum is shifted ≈50 nm. The latter may be explained by internal filter effects caused by considerably higher absorbance of samples used in the 2PA study (see the SI for further experimental details). Close to the region, dashed diagonal line indicating λem = λex, shows two features at 430 and 470 nm. We attribute these shifts of ~10 nm to redshifted versions of the 420 and 460 nm origins, respectively, as discussed above.
These redshifts may arise from an internal filter effect or, in part, a slight shift in the solvent (higher) polarity. Although the exact nature of the associated excited electronic states remains under discussion, it is remarkable that 2-photon excitation produces a picture that is, in many respects, similar to that observed for 1-photon excitation. In particular, the appearance of an origin in the 2-photon spectral map indicates that the excited species most likely lack inversion center(s) because, otherwise, the emission would emanate from a different excited state. Figure 6d summarizes the absorption and emission characteristics of XDDcoTh.
Modeling studies
One reasonable conclusion from the 2PA results is that the two emitting states indicate two forms of conjugation. As noted above, our original work with phenyl SQs indicates conjugation via a cage-centered LUMO; however, our finding of conjugation in LL SQs that had no cages points to another mechanism. In several of our previous papers, we described “unconventional conjugation” in these systems; however, traditional modeling approaches did not indicate the reason for this behavior. Figures S33, S34 represent a traditional Guassian 16 approach to modeling the XDD polymers, which indicate aromatic-centered HOMO and LUMO interactions without conjugation or prediction of the photophysical properties that reflect these interactions.
As with our previous publications [41, 49], examples of calculated HOMOs and LUMOs for the XDD-coTerPh and coTh systems and corresponding absorption spectra presented in Fig. S34 indicate localization of the aromatic components, in contrast to the above evidence for extended conjugation. This result is consistent with the calculated emission behavior (Table S3). Efforts to establish more encompassing modeling approaches are ongoing.
Fortunately, we were able to use DFT studies (see supporting information) to evaluate the structural properties and related bonding phenomena of silicon cage structures coupled with various (hetero)aromatic functional groups connected via disiloxane-vinylene bridges. The optimized structures were assessed to investigate the optoelectronic properties by extending the π-conjugation from n = 1, 2, and 3 repeat units.
Quantum chemical calculations that predict photophysical properties induced by the SiO-bridged cage framework are potentially quite intriguing for the following reasons:
-
(1)
Differences in the electronegativities of Si (1.7) and O (3.5) induce substantial polarization;
-
(2)
Near alignment of orbital energies and symmetries between Si and O result in effective overlap, resulting in energy level variations, optical properties, and molecular packing patterns [50, 51].
-
(3)
A natural bond orbital (NBO) analysis reveal that the O and Si atoms in the cage framework interacted via sp and sp3 hybridization, respectively.
-
(4)
In addition, a partial contribution of hybridization ranging from 2.63 to 3.07% arose from the vacant d-orbitals of the silicon atom (Table S4).
Closer observation of the oxygen lone pair electrons reveal remarkable hybridization of the Si atoms, an indication of possible lateral π-overlap between Si and O atoms [12]. In addition to the existence of Si-O σ-bonds along the x-axis, symmetry matching between the oxygen lone electron pair pz orbitals and two dxy and dxz lobes of Si orbitals lying on either side of the σ-bond led to lateral dπ-pπ interactions (Fig. 7). The empty d-orbitals may have accepted lone pair π-electrons from the pz orbitals of the O-atom, leading to substantial polarization of Siδ+and Oδ− (Table S5). The strengths of the dπ-pπ interactions were not greater than those of pπ-pπ overlap, implying that modest double-bond character may have tuned the optoelectronic properties through their limited π-conjugation pathway. The lengths of the Si-O bonds are predicted to range from 1.632–1.656 Å (Table S6).

a Optimized geometry of the silicon cage framework used for the study. Hydrogen atoms were omitted for clarity. b Schematic illustration of dative π-bonding occurring through pπ-dπ orbital interactions between vacant d-orbitals of silicon and lone pair electrons of oxygen atoms
Energy level variations and electron density distributions in the frontier molecular orbitals were analyzed to understand the structural and electronic properties of the materials. Figure 8 shows that the HOMO and LUMO of (XDDcoStil)1 are computed to be −5.415 and −1.725 eV, respectively, with a band gap of 3.69 eV.

Schematic diagram illustrating the energy level and isodensity surfaces of the frontier molecular orbitals (FMOs) for the π-extended (XDDcoStil)n=1, 2, 3
Figure S
Extending the π-conjugation to n = 2 decrease the band gap via synergistic destabilization and stabilization of the HOMO and LUMO, respectively. However, the band gap reduction of 3.295 eV for n = 3 originates mainly from HOMO destabilization, while the LUMO is unchanged. Identical trends in energy level fluctuations are also found for other derivatives. (Figs. S35–S38).
The major contribution to HOMO destabilization and band gap reduction originates from delocalization over the π-framework through σ*-π* and dπ-pπ orbital overlap interactions [10]. The band gaps obtained for the (XDDcoAr)3 derivatives increase in the order (XDDcoStil)3 (3.295 eV) < (XDDcoTh)3 (3.754 eV) < (XDDcoTerPh)3 (3.971 eV) < (XDDcoBiPh)3 (4.044 eV) < (XDDcoPh)3 (4.059 eV). The (XDDcoStil)3 band gap was the lowest in this series due to extended π-conjugation. The planarity of the thiophene units put (XDDcoTh)3 in second place. However, the dominant ortho-hydrogen effects in biphenyl and terphenyl units marginally affect the band gap.
The electron populations for the HOMOs and LUMOs are distributed within the conjugated (hetero)aromatic segments. The dipole moments computed for the transient excited states are lower than those of the ground state. MESP analysis demonstrates that electropositivity in the cage framework is induced by the Si atoms and further π extension to n = 2 and 3 significantly neutralize the charge populations of the derivatives (Figs. S39–S41).
The spectra for the (XDDcoAr)n derivatives simulated at the M062X/6-311 G**/C-PCM(CH2Cl2) level of theory exhibit a trend consistent with the experimental absorption spectra (Figs. S42, S43). Extending the π-conjugation led to a bathochromic shift in the absorption spectrum with a concomitant increase in the oscillator strength (f). Moreover, the major transitions for (XDDcoAr)n= 1, 2 involved the HOMO and LUMO, which were ascribed to charge transfer (CT) transitions; however, further extension to the n = 3 unit changed the transitions to localized π-π* type, in which the HOMO-1 and LUMO + 1 were major contributors. This trend was corroborated by the DOS-PDOS analysis (Figures S43, S44).
Conclusions
In all of our earlier studies [37, 42, 43, 52,53,54,55], efforts centered on the study of functionalized phenyl and vinyl silsesquioxanes repeatedly demonstrated 3-D conjugation based on the formation of a cage-centered LUMO. More recently, we explored double-decker silsesquioxane copolymers again and found conjugation, but apparently through siloxane endcaps [40, 41, 44]. Most recently, we examined the photophysical properties of ladder copolymers without cages and did not anticipate conjugation given that no cages were involved; thus, no cage-centered LUMOs could have formed [41]. This form of conjugation was termed unconventional because, at the time, there was no logical explanation for why conjugation was observe.
Herein, we continued to attempt to define the extent and source of unexpected conjugation in siloxane and SQ compounds linked by VinylMe2Si-O-SiMe2Vinyl tethers, and we explored unique configurations to identify limiting structures; again, we find conjugation. In the examples presented here, we identified conjugation by redshifted emissions in both the 1PA and 2PA studies, by observing charge transfer from polymeric species to the electron acceptor F4TCNQ, by correlating the degree of polymerization (DP) with the extent of the redshifts and finally with modeling studies that revealed unexpected dπ-pπ conjugations. We also find unexpected failures of Kasha’s rule, in that many of the compounds studied exhibited multiple emitting states, an uncommon feature of conjugated polymers.
In the structures studied, we still could not distinguish between what might be called trans vs. cis conjugation in the structures shown in Scheme 2. A follow on paper will describe conjugation even in simple divinylsiloxane copolymers; this contrasts with what is known about polysiloxanes but supports a second mechanism for conjugation in polysiloxane copolymers [56].

Possible conjugation pathways for XDD copolymers; trans vs. cis or both?
As noted by one of the reviewers, there may be an alternate explanation in which conjugation arises in part from the interactions of oxygen lone electron pairs with silicon σ* orbitals, as described previously [9, 10]. This may also explain the conjugation illustrated in Scheme 3 for the copolymers, which were synthesized to probe the exact nature of this new form of conjugation. The properties of these new systems will be described at a later date, together with more complete theoretical modeling studies of both the model compounds and the complementary copolymers [56, 57].

References
West R. Multiple bonds to silicon: 20 years later. Polyhedron. 2002;21:467–72. https://doi.org/10.1016/S0277-5387(01)01017-8
Raabe G, Michl J. Multiple bonding to silicon. Chem Rev. 1985;85:419–509. https://doi.org/10.1021/cr00069a005
Baceiredo A, Kato T. Multiple bonds to silicon (recent advances in the chemistry of silicon containing multiple bonds). In Organosilicon Compounds; Elsevier, 2017; pp 533–618. https://doi.org/10.1016/B978-0-12-801981-8.00009-5
Boudin A, Cerveau G, Chuit C, Corriu RJP, Reye C. Reactivity of dianionic hexacoordinated silicon complexes toward nucleophiles: a new route to organosilanes from silica. Organometallics. 1988;7:1165–71. https://doi.org/10.1021/om00095a023
Laine RM, Blohowiak KY, Robinson TR, Hoppe ML, Nardi P, Kampf J, Uhm J. Synthesis of pentacoordinate silicon complexes from SiO2. Nature. 1991;353:642–4. https://doi.org/10.1038/353642a0
Chuit C, Corriu RJP, Reye C, Young JC. Reactivity of penta- and hexacoordinate silicon compounds and their role as reaction intermediates. Chem Rev. 1993;93:1371–448. https://doi.org/10.1021/cr00020a003
Kost D, Kalikhman I. Hypercoordinate silicon complexes based on hydrazide ligands. A remarkably flexible molecular system. Acc Chem Res. 2009;42:303–14. https://doi.org/10.1021/ar800151k
Kocher N, Henn J, Gostevskii B, Kost D, Kalikhman I, Engels B, Stalke D. Si−E (E = N, O, F) bonding in a hexacoordinated silicon complex: new facts from experimental and theoretical charge density studies. J Am Chem Soc. 2004;126:5563–8. https://doi.org/10.1021/ja038459r
Fujimoto H, Yabuki T, Tamao K, Fukui K. A theoretical study of chemical bonds in silicon species. J Mol Struct. 1992;260:47–61. https://doi.org/10.1016/0166-1280(92)87034-W
Yamaguchi S, Tamao K. Silole-containing σ- and π-conjugated compounds. J Chem Soc Dalton Trans. 1998, 3693–702. https://doi.org/10.1039/a804491k
Kumar VB, Leitao EM. Properties and applications of polysilanes. Appl Organo Chem. 2020;34:e5402 https://doi.org/10.1002/aoc.5402
Qin Y, Chen H, Yao J, Zhou Y, Cho Y, Zhu Y, Qiu B, Ju C-W, Zhang Z-G, He F, Yang C, Li Y, Zhao D. Silicon and oxygen synergistic effects for the discovery of new high-performance nonfullerene acceptors. Nat Commun. 2020;11:5814 https://doi.org/10.1038/s41467-020-19605-z
Chen J, Cao Y. Silole‐containing polymers: chemistry and optoelectronic properties. Macromol Rapid Commun. 2007;28:1714–42. https://doi.org/10.1002/marc.200700326
Voronkov MG, Lavrent’yev VI. Polyhedral Oligosilsesquioxanes and Their Homo Derivatives. In Inorganic Ring Systems; Boschke FL, Dewar MJS, Dunitz JD, Hafner K, Heilbronner E, Itô S, Lehn J-M, Niedenzu K, Raymond KN, Rees CW, Schäfer K, Vögtle F, Wittig G, Series Eds.; Topics in Current Chemistry; Springer Berlin Heidelberg: Berlin, Heidelberg, 1982; Vol. 102, pp 199–236. https://doi.org/10.1007/3-540-11345-2_12
Schwab JJ, Lichtenhan JD, Chaffee KP, Mather PT, Romo-Uribe A. Polyhedral oligomeric silsesquioxanes (poss): silicon based monomers and their use in the preparation of hybrid polyurethanes. MRS Proc. 1998;519:21 https://doi.org/10.1557/PROC-519-21
Baney RH, Itoh M, Sakakibara A, Suzuki T. Silsesquioxanes. Chem Rev 1995;95:1409–30. https://doi.org/10.1021/cr00037a012
Calzaferri GS. In Tailor-made Silicon-Oxygen Compounds; Friedr. Vieweg & SohnmbH, 1996; pp 149-69.
Lichtenhan J. Silsesquioxane-based polymers. In Polymeric Materials Encyc.; CRC Press, N.Y, 1996; Vol. 10, pp 7768–77.
Provatas A, Matisons JG. Synthesis and applications of silsesquioxanes. In. Trends Polym Sci 1997;5:327–322.
Li G, Wang L, Ni H, Pittman CU,Jr. Polyhedral oligomeric silsesquioxane (POSS) polymers and copolymers: a review. J Inorg Organomet Polym. 2001;11:123–54. https://doi.org/10.1023/A:1015287910502
Duchateau R. Incompletely condensed silsesquioxanes: versatile tools in developing silica-supported olefin polymerization catalysts. Chem Rev 2002;102:3525–42. https://doi.org/10.1021/cr010386b
Abe Y, Gunji T. Oligo- and polysiloxanes. Prog Polym Sci. 2004;29:149–82. https://doi.org/10.1016/j.progpolymsci.2003.08.003
Phillips SH, Haddad TS, Tomczak SJ. Developments in nanoscience: polyhedral oligomeric silsesquioxane (POSS)-polymers. Curr Opin Solid State Mater Sci. 2004;8:21–29. https://doi.org/10.1016/j.cossms.2004.03.002
Kannan RY, Salacinski HJ, Butler PE, Seifalian AM. Polyhedral oligomeric silsesquioxane nanocomposites: the next generation material for biomedical applications. Acc Chem Res. 2005;38:879–84. https://doi.org/10.1021/ar050055b
Laine RM. Nanobuilding blocks based on the [OSiO1.5]x (x = 6, 8, 10) octasilsesquioxanes. J Mater Chem. 2005;15:3725. https://doi.org/10.1039/b506815k
Lickiss PD, Rataboul F. Fully condensed polyhedral oligosilsesquioxanes (POSS): from synthesis to application. Adv Organomet Chem. 2008;57:1–116. https://doi.org/10.1016/S0065-3055(08)00001-4
Chan KL, Sonar P, Sellinger A. Cubic silsesquioxanes for use in solution processable organic light emitting diodes (OLED). J Mater Chem. 2009;19:9103 https://doi.org/10.1039/b909234j
Wu J, Mather PT. POSS polymers: physical properties and biomaterials applications. Polym Rev. 2009;49:25–63. https://doi.org/10.1080/15583720802656237
Cordes DB, Lickiss PD, Rataboul F. Recent developments in the chemistry of cubic polyhedral oligosilsesquioxanes. Chem Rev. 2010;110:2081–173. https://doi.org/10.1021/cr900201r
Laine RM, Roll MF. Polyhedral Phenylsilsesquioxanes. Macromolecules. 2011;44:1073–109. https://doi.org/10.1021/ma102360t
Applications of Polyhedral Oligomeric Silsesquioxanes; Hartmann-Thompson, C., Ed.; Advances in silicon science; Springer: Dordrecht, 2011.
McCabe C, Glotzer SC, Kieffer J, Neurock M, Cummings PT. Multiscale simulation of the synthesis, assembly and properties of nanostructured organic/inorganic hybrid materials. J Comput Theor Nanosci. 2004;1:265–79. https://doi.org/10.1166/jctn.2004.024
Ionescu TC, Qi F, McCabe C, Striolo A, Kieffer J, Cummings PT. Evaluation of force fields for molecular simulation of polyhedral oligomeric silsesquioxanes. J Phys Chem B. 2006;110:2502–10. https://doi.org/10.1021/jp052707j
Bassindale AR, Pourny M, Taylor PG, Hursthouse MB, Light ME. Fluoride-ion encapsulation within a silsesquioxane cage. Angew Chem Int Ed 2003;42:3488–90. https://doi.org/10.1002/anie.200351249
Anderson SE, Bodzin DJ, Haddad TS, Boatz JA, Mabry JM, Mitchell C, Bowers MT. Structural investigation of encapsulated fluoride in polyhedral oligomeric silsesquioxane cages using ion mobility mass spectrometry and molecular mechanics. Chem Mater 2008;20:4299–309. https://doi.org/10.1021/cm800058z
Guan J, Tomobe K, Madu I, Goodson T III, Makhal K, Trinh MT, et al. Photophysical properties of partially functionalized phenylsilsesquioxane: [RSiO1.5]7[Me/nPrSiO1.5] and [RSiO1.5]7[O0.5SiMe3]3 (R = 4-Me/4-CN-Stilbene). Cage-centered magnetic fields form under intense laser light. Macromolecules. 2019;52:4008–19. https://doi.org/10.1021/acs.macromol.9b00699
Laine RM, Sulaiman S, Brick C, Roll M, Tamaki R, Asuncion MZ, Neurock M, Filhol J-S, Lee C-Y, Zhang J, Goodson T, Ronchi M, Pizzotti M, Rand SC, Li Y. Synthesis and photophysical properties of stilbeneoctasilsesquioxanes. emission behavior coupled with theoretical modeling studies suggest a 3-d excited state involving the silica core. J Am Chem Soc 2010;132:3708–22. https://doi.org/10.1021/ja9087709
Guan J, Tomobe K, Madu I, Goodson T, Makhal K, Trinh MT, et al. Photophysical Properties of Functionalized Double Decker Phenylsilsesquioxane Macromonomers: [PhSiO1.5]8[OSiMe2)2 and [PhSiO1.5]8(O0.5SiMe3)4. Cage-Centered Lowest Unoccupied Molecular Orbitals Form Even When Two Cage Edge Bridges Are Removed, Verified by Modeling and Ultrafast Magnetic Light Scattering Experiments. Macromolecules. 2019;52:7413–22. https://doi.org/10.1021/acs.macromol.9b00700
Guan J, Arias JJR, Tomobe K, Ansari R, Marques MdeFV, Rebane A, et al. Unconventional Conjugation via vinylMeSi(O−)2 Siloxane Bridges May Imbue Semiconducting Properties in [Vinyl(Me)SiO(PhSiO1.5)8OSi(Me)Vinyl-Ar] Double-Decker Copolymers. ACS Appl Polym Mater 2020;2:3894–907. https://doi.org/10.1021/acsapm.0c00591
Guan J, Arias JJR, Tomobe K, Ansari R, Marques M de FV, Rebane A, et al. Unconventional Conjugation via vinylMeSi(O−)2 Siloxane Bridges May Imbue Semiconducting Properties in [Vinyl(Me)SiO(PhSiO1.5)8 OSi(Me)Vinyl-Ar] Double-Decker Copolymers. ACS Appl Polym Mater 2020, acsapm.0c00591. https://doi.org/10.1021/acsapm.0c00591
Guan J, Sun Z, Ansari R, Liu Y, Endo A, Unno M, Ouali A, Mahbub S, Furgal JC, Yodsin N, Jungsuttiwong S, Hashemi D, Kieffer J, Laine RM. Conjugated copolymers that shouldn’t be. Angew Chem Int Ed. 2021;60:11115–9. https://doi.org/10.1002/anie.202014932
Asuncion MZ, Laine RM. Fluoride rearrangement reactions of polyphenyl- and polyvinylsilsesquioxanes as a facile route to mixed functional phenyl, Vinyl T 10 and T 12 silsesquioxanes. J Am Chem Soc. 2010;132:3723–36. https://doi.org/10.1021/ja9087743
Jung JH, Furgal JC, Clark S, Schwartz M, Chou K, Laine RM. Beads on a Chain (BoC) Polymers with Model Dendronized Beads. Copolymerization of [(4-NH2PhSiO1.5)6(IPhSiO1.5)2] and [(4-CH3OPhSiO1.5)6(IPhSiO1.5)2] with 1,4-Diethynylbenzene (DEB) Gives Through-Chain, Extended 3-D Conjugation in the Excited State That Is an Average of the Corresponding Homopolymers. Macromolecules. 2013;46:7580–90. https://doi.org/10.1021/ma401422t
Zhang Z, Guan J, Ansari R, Kieffer J, Yodsin N, Jungsuttiwong S, et al. Further proof of unconventional conjugation via disiloxane bonds: double decker sesquioxane [vinylMeSi(O0.5)2(PhSiO1.5)8(O0.5)2SiMevinyl] derived alternating terpolymers give excited-state conjugation averaging that of the corresponding copolymers. Macromolecules. 2022, acs.macromol.2c01355. https://doi.org/10.1021/acs.macromol.2c01355
Liu Y, Takeda N, Ouali A, Unno M. Synthesis, characterization, and functionalization of tetrafunctional double-decker siloxanes. Inorg Chem 2019;58:4093–8. https://doi.org/10.1021/acs.inorgchem.9b00416
Endo H, Takeda N, Unno M. Synthesis and properties of phenylsilsesquioxanes with ladder and double-decker structures. Organometallics. 2014;33:4148–51. https://doi.org/10.1021/om500010y
Demchenko AP, Tomin VI, Chou P-T. Breaking the Kasha rule for more efficient photochemistry. Chem Rev. 2017;117:13353–81. https://doi.org/10.1021/acs.chemrev.7b00110
del Valle JC, Catalán J. Kasha’s rule: a reappraisal. Phys Chem Chem Phys. 2019;21:10061–9. https://doi.org/10.1039/C9CP00739C
Guan J, Tomobe K, Madu I, Goodson T, Makhal K, Trinh MT, et al. Photophysical Properties of Partially Functionalized Phenylsilsesquioxane: [RSiO1.5]7[Me/nPrSiO1.5] and [RSiO1.5]7[O0.5SiMe3]3 (R = 4-Me/4-CN-Stilbene). Cage-Centered Magnetic Fields Form under Intense Laser Light. Macromolecules. 2019;52:4008–19. https://doi.org/10.1021/acs.macromol.9b00699
Dankert F, Hänisch C. Siloxane coordination revisited: Si−O bond character, reactivity and magnificent molecular shapes. Eur J Inorg Chem. 2021;2021:2907–27. https://doi.org/10.1002/ejic.202100275
Fugel M, Hesse MF, Pal R, Beckmann J, Jayatilaka D, Turner MJ, Karton A, Bultinck P, Chandler GS, Grabowsky S. Covalency and ionicity do not oppose each other—relationship between Si−O bond character and basicity of siloxanes. Chem Eur J. 2018;24:15275–86. https://doi.org/10.1002/chem.201802197
Sulaiman S, Bhaskar A, Zhang J, Guda R, Goodson T, Laine RM. Molecules with perfect cubic symmetry as nanobuilding blocks for 3-D assemblies. elaboration of octavinylsilsesquioxane. Unusual luminescence shifts may indicate extended conjugation involving the silsesquioxane core. Chem Mater. 2008;20:5563–73. https://doi.org/10.1021/cm801017e
Sulaiman S, Zhang J, Goodson T III, Laine RM. Synthesis, characterization and photophysical properties of polyfunctional phenylsilsesquioxanes: [O-RPhSiO1.5]8, [2,5-R2PhSiO1.5]8, and [R3PhSiO1.5]8. Compounds with the highest number of functional units/unit volume. J Mater Chem. 2011;21:11177. https://doi.org/10.1039/c1jm11701g
Furgal JC, Jung JH, Clark S, Goodson T, Laine RM. Beads on a chain (BoC) phenylsilsesquioxane (SQ) polymers via F − catalyzed rearrangements and ADMET or reverse heck cross-coupling reactions: through chain, extended conjugation in 3-D with potential for dendronization. Macromolecules. 2013;46:7591–604. https://doi.org/10.1021/ma401423f
Bahrami M, Hashemi H, Ma X, Kieffer J, Laine RM. Why Do the [PhSiO 1.58,10,12> Cages self-brominate primarily in the ortho position? modeling reveals a strong cage influence on the mechanism. Phys Chem Chem Phys. 2014;16:25760–4. https://doi.org/10.1039/C4CP03997A
Zhang Z, Kaehr H, Laine RM. Polysiloxane copolymers demonstrate conjugation through Si-O-Si bonds, 2023.
Z Zhang; JJR Arias; H Kaehr,; Y Liu; M Takahashi; R Murata, et al. Conjugation through Si-O-Si bonds, extended examples via SiO0.5/SiO1.5 units. Multiple emissive states in violation of Kasha’s rule., TBD.
Acknowledgements
The Laine and Rebane groups gratefully thank NSF Chemistry for the collaborative research award No. 1610344. Support from the Estonian National Science Foundation grant PRG661 is acknowledged (Ramo and Rebane). The Unno/Liu group is grateful for support from the NEDO project (JPNP06046). Professor Jungsuttiwong thanks NSRF via the Program Management Unit for Human Resources & Institutional Development, Research and Innovation [B16F640099] for funding work performed by her team. The work performed at The Georgia Institute of Technology was made possible through the Air Force Office of Scientific Research (AFOSR) under support provided by the Organic Materials Chemistry Program (Grant FA9550-20-1-0353, Program Manager: Dr. Kenneth Caster).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Arias, J.J.R., Zhang, Z., Takahashi, M. et al. Conjugation in polysiloxane copolymers via unexpected Si-O-Si dπ-pπ overlap, a second mechanism?. Polym J (2024). https://doi.org/10.1038/s41428-024-00899-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41428-024-00899-5
