
Abstract
HEAT repeats are 37–47 amino acid flexible tandem repeat structural motifs occurring in a wide variety of eukaryotic proteins with diverse functions. Due to their ability to undergo elastic conformational changes, they often serve as scaffolds at sites of protein interactions. Here, we describe four affected children from two families presenting with pontocerebellar hypoplasia manifest clinically with neonatal seizures, severe intellectual disability, and motor delay. Whole exome sequencing identified biallelic variants at predicted splice sites in intron 31 of HEATR5B, encoding the HEAT repeat-containing protein 5B segregating in a recessive fashion. Aberrant splicing was found in patient fibroblasts, which correlated with reduced levels of HEATR5B protein. HEATR5B is expressed during brain development in human, and we failed to recover live-born homozygous Heatr5b knockout mice. Taken together, our results implicate loss of HEATR5B in pontocerebellar hypoplasia.
Similar content being viewed by others

Introduction
Pontocerebellar hypoplasia (PCH) refers to a group of recessive neurodevelopmental disorders characterized by loss of parenchymal volume of the pons and cerebellum. PCH frequently follows a degenerative course, with clinical hallmarks including defects in brainstem and cerebellar function, evident by difficulty with swallowing, oculomotor defects, spastic quadriparesis, epilepsy, frequent pulmonary infections, and early death [1]. Currently, there are 11 overlapping clinical subtypes and 17 PCH-related genes that have been described, implicating genes involved in RNA processing or protein translation [2].
HEAT (Huntingtin, Elongation factor 3, the PR65/A subunit of protein phosphatase 2A, and TOR) repeats (HEATRs) are 37–47 amino acid motifs consisting of duplicated anti-parallel α-helices linked by flexible inter-unit loops [3]. Structurally related to armadillo repeats, HEATRs occur in a variety of eukaryotic proteins mediating protein interactions involved in processes such as cytoplasmic and nuclear transport, microtubule dynamics, or chromosome segregation [4]. Upon interaction with binding partners HEATRs are capable of undergoing flexible and elastic changes [5]. Although HEAT repeats occur in dozens of proteins, a total of ten different genes are designated with the name HEATR in the human genome (HEATR 1, 2, 3, 4, 5A, 5B, 6, 7A, 7B1, and 8), and none yet have been linked to human disease, except in a single family with primary ciliary dyskinesia with a homozygous HEATR2 missense allele (CILD18; MIM: 614874) [6].
Materials and methods
Patient recruitment
The Institutional Review Board at the University of California, San Diego, approved this study. All study participants signed informed consent documents, and the study was performed in accordance with Health Insurance Portability and Accountability Act Privacy Rules. The procedures followed for recruitment and data collection were in accordance with the ethical standards of the responsible committee on human experimentation at the respective, participating institute and proper informed consent was obtained.
DNA extraction and whole exome sequencing
DNA was extracted on an Autopure LS instrument (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Samples of the three affected individuals and their unaffected parents were subjected to Agilent Sure-Select Human All Exon v2.0 (44 Mb baited target) library preparation sequencing on an Illumina HiSeq 2000 with v2 chemistry (Read Length: 151). The accession number for these data is dbGAP: phs000288.v1.p1. The Genome Analysis Toolkit workflow was used to identify variants that were homozygous in the children [7].
Computational analysis
Variant calling and filtering were performed according to an established whole exome sequencing (WES) pipeline [7]. Runs of homozygosity were assessed using HomozygosityMapper [8,9,10]. Variants were filtered if not consistent with recessive monogenic inheritance and if the minor allele frequency was >1:10,000 in gnomAD or >1:1000 in our in-house ethnically matched exome database consisting of 7000 sequenced individuals. Missense variants were prioritized by their conservation scores in GERP++ and PhyloP and deleterious or pathogenic scores in at least two of the following in silico pathogenicity prediction tools: Combined Annotation Dependent Depletion, MutationTaster, Polyphen-2, and SIFT.
Reverse transcription (RT) PCR
Patient fibroblasts for Family 2610 were harvested from a skin dermal punch biopsy as described [11]. Total RNA from quantified patient and control fibroblasts was reverse-transcribed using the Superscript III First-Strand cDNA Kit (Invitrogen, Cat: 18080051). PCR analysis of cDNA was designed against exons 22–23 (hHEATR5B_ex22–23_F; hHEATR5B_ex22–23_R, Supplementary Table S1), 30–31 (hHEATR5B_ex30–31_F; hHEATR5B_ex30–31_R, Supplementary Table S1), and 33–34 (hHEATR5B_ex33–34_F, hHEATR5B_ex33–34_R, Supplementary Table S1) of HEATR5B, excluding introns using GoTaq Master Mix (Promega, Cat: M7833). Quantification of band intensity was performed using ImageJ software and normalized to loading control.
Gel extraction
DNA was purified from agarose gels following RT-PCR and recovered using the Zymoclean Gel DNA Recovery Kit as per manufacturer’s instructions (Zymo Research, Cat: D4001).
Western blot
Cells were lysed with ice-cold RIPA buffer (150 mM sodium chloride, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris pH 8.0) and assessed by western blot using standard protocols. Primary antibodies used were anti-beta-Actin (Santa Cruz, sc-47778, 1:2000), and anti-HEATR5B (Sigma-Aldrich, HPA055639, 1:200). Western blot signal intensity (area of peaks) was analyzed using ImageJ and normalized to the loading control for quantification. N = 3 independent extractions used for quantification.
Mouse
Animal use followed NIH guidelines and was approved by IACUC at the University of California, San Diego. The guide 5′-GGATTGTTCATTGCTCTCTT-3′ was co-injected with recombinant CRISPR/Cas9 into C57Bl/6 zygotes according to standard protocols at the UCSD Transgenic Mouse Core. We recovered several damaging alleles and focused on a Heatr5b frameshift allele in constitutively spliced exon 28 of 36 (Refseq (NM_001081179.1): c.4385dupA; p.Asp1462GlufsTer15). Mice were genotyped by PCR followed by Sanger sequencing (mHeatr5b_F/R; Supplementary Table S1). Heterozygous mice were backcrossed to wildtype C57Bl/6 for at least five generations prior to intercross of heterozygotes to generate homozygous animal. Timed pregnant animals were obtained by plug checks, where the day of the observed vaginal plug was determined to be E0.5.
Statistical analysis
Statistical analysis was done as indicated in figure legends using GraphPad Prism 7. Specific tests and p values are indicated for each figure (Figs. 2 and 3). Visualization of data was performed using GraphPad Prism 7.
Results
Clinical evaluation of four affected individuals from two families with similar neurological phenotypes
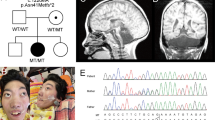
We identified four affected members from two unrelated families with similar neurological phenotypes. Family 2610 had documented parental consanguinity and presented with two affected children (II-1 and II-2) with severe global motor and cognitive delays and frequent neonatal-onset seizures (Fig. 1 and Table 1). The older sibling was born at 36 weeks of gestation and showed intractable convulsions and hypocalcemia in the neonatal period. Absence of meconium led to a diagnosis of aganglionic megacolon that required surgery at 11 days of age. Physical examination showed growth retardation, severe intellectual disability, and gross motor delay with absent head control. There were also microphthalmia, low set ears, talipes equinovarus, increased deep tendon reflexes, and limb spasticity. The younger sibling showed similar features, without megacolon, but instead showed transient polycythemia. Brain MRI for both children revealed pontocerebellar and corpus callosum hypoplasia with a greater degree of ventriculomegaly in the older child, suggesting a progressive course. The older sibling passed away at 3 years of age, while the younger sibling is still living.

A, B Family pedigrees for Families 2610 and 3195. Hash line: deceased. Triangle: deceased pregnancy at 1.5 months gestation. Double line: reported consanguinity. C–F MRIs for individuals 2610-II-2 (C, D) and 3195-III-4 (E, F). Shown are midline sagittal (C, E) and coronal (D, F) images. Asterisk: mega cisterna magna. White arrow: cerebellar hypoplasia. Green arrow: pontine hypoplasia. G HEATR5B 36 exons with genomic position. Red lines: position of intron 31 pathogenic variants (RefSeq NC_000002.12 (NM_019024.2)). Exons were numbered using the UCSC Genome Browser as a reference (uc002rpp.1). H Schematic of the HEATR5B protein with location of the three HEAT repeat domains (RefSeq NP_061897.1). Red dashed line highlights the exon 31/32 boundary at amino acids 1682–1683, between which the two splicing variants were present. aa amino acid.
Family 3195 had two affected children (III-3 and III-4) and two healthy siblings from a first-cousin marriage. Both children had severe global motor and cognitive delay with seizure onset at 3 and 7 months of age, respectively. Brain MRI for the younger sibling revealed PCH, defective myelination, mild cortical atrophy, corpus callosum hypoplasia, and mild ventriculomegaly. Both children passed away at 4 years of age. DNA or MRIs for one of the affected children (III-4) could not be obtained; however, phenotypic similarity between the two siblings suggested a single genetic etiology.
Homozygous HEATR5B damaging variants in pontocerebellar hypoplasia
Following informed consent from all participating individuals (or their guardians), WES was performed on blood-derived DNA from the father, mother, and both affected children of Family 2610, as well as the father, mother, and the affected girl from Family 3195. Rare, potentially deleterious variants were prioritized using an in-house exome database consisting of over 5000 ethnically matched individuals, in addition to publicly available exome datasets, cumulatively numbering over 10,000 individuals [12, 13]. In Family 2610, only one candidate passed filters and segregated with the phenotype, which we identified as a homozygous variant predicted to affected splicing in HEATR5B (Supplementary Table S2). In Family 3195, the variant in HEATR5B was the only splicing variant we identified, among additional missense variants in other genes (Supplementary Table S2). Thus, both families showed homozygous variants adjacent to canonical splice sites (c.5051–1G>A for Family 2610 and c.5050+4A>G for Family 3195) in intron 31 of HEATR5B (RefSeq NC_000002.12 (NM_019024.2)), predicted to impair splicing.
Neither HEATR5B variant was present in the gnomAD database, or in our in-house exome database. The predicted genotypes from next-generation sequencing were confirmed through Sanger sequencing for all individuals where DNA was available (Supplementary Fig. S1). Furthermore, both variants were adjacent to or within a stretch of extended homozygosity (HomozygosityMapper, Supplementary Fig. S2), and were predicted as “disease-causing” by MutationTaster [8,9,10]. HEATR5B is ubiquitously expressed in adult human tissues (GTEx data) and during human brain development (BrainSpan data) at all assessed stages (Supplementary Figs. S3 and S4). The genetic support, the similar location of the variants, as well as the matching phenotypes rendered the HEATR5B variants as the most likely cause of disease.
Pathogenic variants in HEATR5B result in aberrant splicing and subsequent reduced protein levels
To functionally assess splicing defects that may result from these variants, we generated dermal fibroblast cultures from the affected girl and both parents in Family 2610 following informed consent. We assessed the presence of HEATR5B mRNA using RT-PCR from exon 22 to 23, which are upstream of the variant in Family 2610, and observed no differences in expression level compared with control (Fig. 2A, D). Primers located in exon 31 and 32, which flank the variant, demonstrated a lower band corresponding to spliced mRNA, which was of decreased intensity in the affected compared with control or parents after normalization of PCR to a GAPDH loading control (P < 0.0001, Fig. 2B, E). In addition, in the affected there was a new upper band, corresponding to a product in which intron 31 failed to splice out of the transcript (P < 0.0001, Fig. 2B, F). Primers located in exon 33 and 34, which are downstream of the variant, showed no difference in expression level. The data suggest that the variant in intron 31 can lead to aberrant splicing of the transcript.

A, C RT-PCR from fibroblasts from unaffected father (F), mother (M), and affected child (A2) from Family 2610, for primers located in exons 22 and 23 or exons 33 and 34. Results show indistinguishable band intensities and molecular weight in all. B RT-PCR for primers located in exons 31 and 32, spanning the variant, showing reduced intensity of wildtype band (lower band) and presence of a novel upper band, suggesting intron retention in A2 (arrowhead). NTC negative template control. D–G Band quantification of RT-PCR analyses. H RT-PCR of sample A2 showing upper and lower bands used for Sanger sequencing. I Sequence trace from the upper band revealed retention of intron 31. J Sequence trace from the lower band revealed a loss of six nucleotides from the start of exon 32, resulting in the predicted loss of two amino acids in the encoded protein, indicating use of the alternative splice site within exon 32. Red asterisk indicates the new splice site location resulting from the variant. K Schematic of HEATR5B pre-mRNA sequence of intron 31 of 35. Black asterisk indicates the location of the canonical splice site affected by the variant in Family 2610. Statistical analysis in D–G was done using a one-way ANOVA and Tukey’s multiple comparisons test. Unless indicated, differences were not significant between the categories. ****P < 0.0001. N = 3 independent experiments and graphs show individual data points as well as mean ± SEM.
We extracted DNA from both the upper and lower bands of the affected individual following RT-PCR. Sequencing of the extracted DNA confirmed intron retention in the upper band (Fig. 2H, I), and showed a loss of six nucleotides (Fig. 2H, J) in the lower band. This suggests that loss of the original splice site resulted in two new splicing events: (1) usage of a weaker splice site six base pairs downstream of the start of exon 32/36 and (2) failure to splice the intron completely. These result in the loss of two amino acids in the final protein, or higher levels of intron retention, respectively.
We next performed western analysis for HEATR5B protein in whole cell lysates from Family 2610 and observed significantly reduced protein in the sample from the affected compared to both parents and an unrelated control (P < 0.0001, Fig. 3A, B).

A Western blot for HEATR5B full-length protein and β-actin using the same sample input. Full-length HEATR5B signal is reduced in the proband. B Band quantification of western blot analyses. Statistical analysis done using a one-way ANOVA and Tukey’s multiple comparisons test. Unless indicated, differences were not significant between the categories. ****P < 0.0001. N = 3 independent experiments and graph shows individual data points as well as mean ± SEM.
Embryonic lethality of Heatr5b mouse model of disease
To investigate the importance of HEATR5B in development, we generated a frameshift allele (Refseq NM_001081179.1; c.4385dupA; p.Asp1462GlufsTer15) in the mouse homolog Heatr5b using CRISPR/Cas9 pronuclear injection (Supplementary Fig. S5). While heterozygous animals were healthy and fertile, we failed to recover any homozygous mutant mice at P0. We thus established timed pregnant matings but also failed to recover any healthy embryos genotyped as homozygous mutants at embryonic day (E) 12.5 or 14.5 (0/21 embryos total; P = 0.0041). These results are consistent with an essential role in embryonic development and supported pathogenicity of HEATR5B function perturbation.
Discussion
Here, we describe two unrelated families with four affected individuals that exhibited a similar phenotypic spectrum comprising severe global motor and developmental delay, PCH, and epilepsy. Both families harbored unique splice site variants in HEATR5B that segregated with the phenotype in affected individuals, likely impaired splicing, and resulted in reduced steady-state levels of the encoded HEATR5B protein in a fibroblast culture. Embryonic lethality of Heatr5b knockout mice further supports the significance of this protein in development. This severe phenotype in the knockout mouse suggests that these patient splice site variants are hypomorphic, which is supported by the residual expression of protein in fibroblasts that are homozygous for one of these variants.
While the cellular role of the HEATR5B protein in mammals remains unclear, recent studies on the yeast homolog of this protein, Laa1, have provided evidence for its importance in endocytosis; this is based on its interaction with the adaptor protein complex-1 (AP-1) [14], a highly conserved clathrin adaptor involved in membrane trafficking [15, 16]. This association of HEATR5 proteins with endocytosis is evolutionarily conserved from yeast to mammals [17,18,19,20]. Although disruption of the HEATR5 family causes strong defects in AP-1 recruitment in all systems tested so far, it remains unclear whether HEATR5B plays a direct role in AP-1 function [17,18,19]. At the minimum, the identification of deleterious splice site variants in patients with PCH and epilepsy suggests that its function is important for mammalian neurodevelopment.
Eleven subtypes of PCH have been described thus far, with two subtypes (PCH 3 and PCH 11) previously implicated in intracellular vesicle transport or vesicle formation [20,21,22,23,24]. Due to the proposed role of HEATR5B in endocytosis, it is possible that this newly associated HEATR5B functions in endocytosis although more work is required to assess function, and there is no obvious convergence on a subtype-specific set of phenotypes, due to the limited number of cases reported in the literature [25].
Web resources
RefSeq, http://www.ncbi.nlm.nih.gov/RefSeq. Online Mendelian Inheritance in Man (OMIM), http://www.omim.org. HomozygosityMapper, http://www.homozygositymapper.org/. MutationTaster, http://www.mutationtaster.org/. Exo-me aggregation Consortium, ExAc Browser http://exac.broadinstitute.org/. GME Variome, http://igm.ucsd.edu/gme. The Genotype-Tissue Expression (GTEx) Project, https://www.gtexportal.org/home/. BrainSpan – Atlas of the Developing Human Brain, http://www.brainspan.org/. SimulConsult – Measurement resources. http://www.simulconsult.com/resources/measurement.html. Python Software Foundation, https://www.python.org/. UniProt Database, http://www.uniprot.org.
Data availability
Data are available in a public, open access repository. The exome sequencing from individuals from the University of California, San Diego, study site have been deposited in the Database of Genotypes and Phenotypes under accession number dbGaP:phs000288.v2.p2. Clinvar accession IDs for this submision are: SCV001468900 and SCV001468901.
References
Rudnik-Schoneborn S, Barth PG, Zerres K. Pontocerebellar hypoplasia. Am J Med Genet C Semin Med Genet. 2014;166C:173–83.
van Dijk T, Baas F, Barth PG, Poll-The BT. What’s new in pontocerebellar hypoplasia? An update on genes and subtypes. Orphanet J Rare Dis. 2018;13:92.
Groves MR, Hanlon N, Turowski P, Hemmings BA, Barford D. The structure of the protein phosphatase 2A PR65/A subunit reveals the conformation of its 15 tandemly repeated HEAT motifs. Cell. 1999;96:99–110.
Neuwald AF, Hirano T. HEAT repeats associated with condensins, cohesins and other complexes involved in chromosome-related functions. Genome Res. 2000;10:1445–52.
Grinthal A, Adamovic I, Weiner B, Karplus M, Kleckner N. PR65, the HEAT-repeat scaffold of phosphatase PP2A, is an elastic connector that links force and catalysis. Proc Natl Acad Sci USA. 2010;107:2467–72.
Horani A, Druley TE, Zariwala MA, Patel AC, Levinson BT, Van Arendonk LG, et al. Whole-exome capture and sequencing identifies HEATR2 mutation as a cause of primary ciliary dyskinesia. Am J Hum Genet. 2012;91:685–93.
Dixon-Salazar TJ, Silhavy JL, Udpa N, Schroth J, Bielas S, Schaffer AE, et al. Exome sequencing can improve diagnosis and alter patient management. Sci Transl Med. 2012;4:138ra178.
Seelow D, Schuelke M, Hildebrandt F, Nurnberg P. HomozygosityMapper—an interactive approach to homozygosity mapping. Nucleic Acids Res. 2009;7:W593–9.
Seelow D, Schuelke M. HomozygosityMapper2012—bridging the gap between homozygosity 291 mapping and deep sequencing. Nucleic Acids Res. 2012;40:W516–520.
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–6.
Villegas J, McPhaul M. Establishment and culture of human skin fibroblasts. Curr Protoc Mol Biol. 2005;28:23.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Scott EM, Halees A, Itan Y, Spencer EG, He Y, Azab MA, et al. Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat Genet. 2016;48:1071–6.
Zysnarski CJ, Lahiri S, Javed FT, Martinez-Marquez JY, Trowbridge JW, Duncan MC. Adaptor protein complex-1 (AP-1) is recruited by the HEATR5 protein Laa1 and its co-factor Laa2 in yeast. J Biol Chem. 2019;4:1410–9.
Canagarajah BJ, Ren X, Bonifacino JS, Hurley JH. The clathrin adaptor complexes as a paradigm for membrane-associated allostery. Protein Sci. 2013;22:517–29.
Traub LM. Common principles in clathrin-mediated sorting at the Golgi and the plasma membrane. Biochim Biophys Acta. 2005;1744:415–37.
Fernández GE, Payne GS. Laa1p, a conserved AP-1 accessory protein important for AP-1 localization in yeast. Mol Biol Cell. 2006;17:3304–17.
Gillard G, Shafaq-Zadah M, Nicolle O, Damaj R, Pecreaux J, Michaux G. Control of E-cadherin apical localisation and morphogenesis by a SOAP-1/AP-1/clathrin pathway in C. elegans epidermal cells. Development. 2015;142:1684–94.
Yu Y, Kita A, Udo M, Katayama Y, Shintani M, Park K, et al. Sip1, a conserved AP-1 accessory protein, is important for Golgi/endosome trafficking in fission yeast. PLoS One. 2012;7:e45324.
Le Bras S, Rondanino C, Kriegel-Taki G, Dussert A, Le Borgne R. Genetic identification of intracellular trafficking regulators involved in Notch-dependent binary cell fate acquisition following asymmetric cell division. J Cell Sci. 2012;125:4886–901.
Ahmed MY, Chioza BA, Rajab A, Schmitz-Abe K, Al-Khayat A, Al-Turki S, et al. Loss of PCLO function underlies pontocerebellar hypoplasia type III. Neurology 2015;17:1745–50.
Durmaz B, Wollnik B, Cogulu O, Li Y, Tekgul H, Hazan F, et al. Pontocerebellar hypoplasia type III (CLAM): extended phenotype and novel molecular findings. J Neurol. 2009;3:416–9.
Ivanova EL, Mau-Them FT, Riazuddin S, Kahrizi K, Laugel V, Schaefer E, et al. Homozygous truncating variants in TBC1D23 cause pontocerebellar hypoplasia and alter cortical development. Am J Hum Genet. 2017;101:428–40.
Marin-Valencia I, Gerondopoulos A, Zaki MS, Ben-Omran T, Almureikhi M, Demir E, et al. Homozygous mutations in TBC1D23 lead to a non-degenerative form of pontocerebellar hypoplasia. Am J Hum Genet. 2017;101:441–50.
van Dijk T, Baas F, Barth PG, Poll-The BT. What’s new in pontocerebellar hypoplasia? An update on genes and subtypes. Orphanet J Rare Dis. 2018;13:92.
Acknowledgements
We are indebted to the families for their participation in this study. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from: the GTEx Portal on March 3, 2018.
Funding
This work was supported by NIH grants R01NS048453 and R01NS052455 (to JGG). SGG was supported by the Ruth L. Kirschstein Institutional National Research Service Award (T32 GM008666) and from the National Institute on Deafness and Other Communication Disorders (F31HD095602). MWB was supported by an EMBO Long-Term Fellowship (ALTF 174–2015), co-funded by the Marie Curie Actions of the European Commission (LTFCOFUND2013, GA-2013–609409), and an Erwin Schrödinger Fellowship by the Austrian Science Fund (FWF, J 4197-B30). The authors thank Broad Institute (U54HG003067 to Eric Lander and UM1HG008900 to D. MacArthur) and the Yale Center for Mendelian Disorders (U54HG006504 to M. Gunel), Center for Inherited Disease Research for genotyping and sequencing support.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Ghosh, S.G., Breuss, M.W., Schlachetzki, Z. et al. Biallelic hypomorphic mutations in HEATR5B, encoding HEAT repeat-containing protein 5B, in a neurological syndrome with pontocerebellar hypoplasia. Eur J Hum Genet 29, 957–964 (2021). https://doi.org/10.1038/s41431-021-00832-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-021-00832-x
This article is cited by
-
Evaluation of the Patients with the Diagnosis of Pontocerebellar Hypoplasia: A Multicenter National Study
The Cerebellum (2024)
-
Genome-wide association study using whole-genome sequencing identifies risk loci for Parkinson’s disease in Chinese population
npj Parkinson's Disease (2023)
-
Clinical genomics—but faster
European Journal of Human Genetics (2021)
