
Abstract
Congenital diaphragmatic hernia (CDH) is a life-threatening malformation characterised by failure of diaphragmatic development with lung hypoplasia and persistent pulmonary hypertension of the newborn (PPHN). The incidence is 1:2000 corresponding to 8% of all major congenital malformations. Morbidity and mortality in affected newborns are very high and at present, there is no precise prenatal or early postnatal prognostication parameter to predict clinical outcome in CDH patients. Most cases occur sporadically, however, genetic causes have long been discussed to explain a proportion of cases. These range from aneuploidy to complex chromosomal aberrations and specific mutations often causing a complex phenotype exhibiting multiple malformations along with CDH. This review summarises the genetic variations which have been observed in syndromic and isolated cases of congenital diaphragmatic hernia.
Similar content being viewed by others
Introduction
Congenital diaphragmatic hernia (CDH), accounting for roughly 8% of all major congenital malformations is a severe physical deformity associated with high morbidity and mortality and occurs in less than 5 cases in 10,000 births [1, 2]. Due to improved treatment options survival has greatly improved up to 88% [3] but long-term morbidity commonly remains an important issue [4]. CDH is caused by a discontinuity in the diaphragm allowing abdominal organs to penetrate into the thoracic cavity, to interfere with heart and lung development thus causing a series of severe pathophysiologic events: pulmonary hypoplasia, pulmonary hypertension (PH) following increased pulmonary vascular resistance (PVR) and cardiac impairment are hallmarks of CDH [5]. Notably, most cases occur sporadically being referred to as isolated or nonsyndromic, respectively. However, genetic causes ranging from aneuploidy to complex chromosomal aberrations and specific mutations have long been discussed to explain a proportion of cases of CDH often along with additional malformations [6, 7]. This review summarises the recurrent genetic variations which have been observed in syndromic and isolated cases of congenital diaphragmatic hernia, including copy number variations, point mutations and the role of vitamin A homoeostasis and signalling pathways.
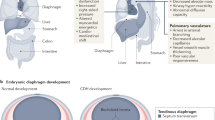
Normal diaphragm and lung development
During embryonic development, the intraembryonic coelom arises as a precursor of the body’s cavities. The cranially located right and left limb of the coelum, referred to as pleuropericardial canals, adhere to the growing lungs as they are pushed away by the growing respiratory organs. Thereby, thin folds appear on the right and left side of the heart: the pleuropericardial folds cranially and the pleuroperitoneal folds caudally. Together with the transverse septum, the esophageal mesentery, and the posthepatic mesenchymal plate (PHMP), the latter orchestrate the formation of the diaphragm [8, 9]. If the pleuroperitoneal folds close insufficiently or if a weakness within the musculature or connective tissue remains, the congenital diaphragmatic hernia can occur. For a detailed explanation on lung and diaphragm development, please refer to the supplemental material.
Genetic variations in congenital diaphragmatic hernia
The significance of copy number variations (CNVs)
Several studies have examined the association between the CNVs and certain diseases and provide evidence for both causative and predisposing relations [10,11,12]. To what extent these genomic imbalances could potentially contribute to CDH development has long been unclear but recent scientific evidence sheds light on this relatively new genetic approach, revealing a subset of as much as 10% of CDH cases as attributable to CNVs [13]. Notably, most CNVs that have so far been identified to play a role in the development of CDH are deletions, which in the context of haploinsufficiency are considered deleterious in the process of diaphragm development. However, several CDH-related chromosomal loci are affected by copy number gains. To date, CNVs putatively responsible for CDH could be found in numerous loci. Zhu et al. compared 196 CDH cases to 987 healthy controls and identified six CNVs significantly associated with CDH (Suppl. Table 1) [14]. Of these, a gain in copy number of parts of HLX1 found in five patients with CDH is easiest to associate to the developing diaphragm since Hlx could be determined in the murine embryonic septum transversum and diaphragm [15, 16]. Also, Hlx-signal was colocalized with Myod in myoblasts of the upper somites [15] suggesting its contribution to myogenesis. These findings make HLX1 an interesting candidate gene in CDH.
Disruption of essential signalling pathways can lead to CDH
Disp1 is a protein similar to the hedgehog-receptor Ptch acting in the Sonic Hedgehog (Shh) signalling pathway [17]. In mouse studies, Disp1 is specifically expressed in the PPFs of the developing premature diaphragm [18]. Yet, Shh-expressing cells were shown to neither contribute to the muscular, nor to the nonmuscular part of the developing murine diaphragm in fate-mapping studies, and Ptch1 was not found in the PPFs [18]. Therefore, the Disp1 function remains unknown in diaphragm development. However, murine Shh-signalling contributes to phrenic nerve outgrowth [18], which in neural tube cells is executed by the transcription factors Gli2 and Gli3 [19] and a promotor element of NR2F2 (COUP-TFII) responsive to Shh-signalling was identified in a murine cell line [20]. A similar activation of NR2F2 during human diaphragm development has yet to be uncovered. But since Shh-signalling is crucial for embryonic lung [21] and phrenic nerve development and mutations of GLI2 and GLI3 are described in human cases of CDH [22, 23], Shh-signalling could still influence the developing human diaphragm. Furthermore, Kantarci et al. discovered two distinct deletions on chromosome 1 encompassing both HLX and DISP1 associated with CDH that consistently remain of uncertain significance [18]. In contrast, there is evidence for functional Wnt/β-catenin-signalling in the developing diaphragm upstream of which acts WT1 to promote β-catenin expression and cell proliferation especially in the posterior diaphragm [24]. In the embryo, canonical Wnt-signalling is an essential driver for development and requires multiple cellular proteins like WNT5A and FZD2 [25]. Consistently, there is evidence suggesting disrupted Wnt-signalling could contribute to CDH in a male infant harbouring a deletion in 17q12.2 encompassing FZD2 [26]. In line with the importance of Wnt/β-catenin-signalling, Scott et al. reported on a male infant exhibiting a deletion on chromosome 11p13 covering WT1 and PAX6 [27]. Notably, PAX6 is required in myogenesis and its contribution to the CDH phenotype could not be excluded.
Low copy repeats at 8p23 are responsible for CNVs leading to CDH
A mutational hotspot for structural chromosomal aberrations (e.g., translocations, inversions) and microaberrations (e.g., point mutations, base deletions, indels) is the short arm of chromosome 8. Yu et al. and Keitges et al. determined CNVs at 8p23.1 in CDH patients with additional ventricular septal defect (VSD), atrial septal defect (ASD) or incomplete atrioventricular canal defect, or both ASD and VSD [28, 29]. In one case, only two genes, namely GATA4 and NEIL2, were affected by the deletion pinpointing on another CDH candidate region located at 8p23. NEIL2 is involved in base excision repair and is therefore not considered a good CDH candidate. At that time, loss of function mutations of GATA4 were already associated to cardiac anomalies [30, 31] and today we know that GATA4 is important for both embryonic cardiac and lung development [32, 33]. Furthermore, the examination of diaphragm development in mice in which Gata4 was specifically deleted in the PPFs identified muscle connective tissue fibroblasts as a source of CDH in Gata4-deficient organisms [34]. Developing CDH can therefore be associated with GATA4 deficiency in the nonmuscular mesenchyme. Additionally, Wat at al. report on a male infant with a heterozygous de novo del(8p23.1) mutation encompassing GATA4, exhibiting multiple cardiac anomalies along with anterior, right-sided CDH [35] similar to those observed in Gata4+/∆ex2 mice [33]. Notably, his monozygotic twin brother presented with cardiac defects only, omitting CDH despite the presence of a similar mutation also involving GATA4 [35]. These findings from animal studies suggest a striking value of GATA4 transcriptional activity in the developing diaphragm. Notably, both deletions encompassed SOX7, another transcription factor that acts upstream of GATA4 and might multiply the disrupting effects of GATA4 deficiency either if SOX7 itself is deleted or affected by missense mutations [36].
NR2F2 (COUP-TFII) – Key element in CDH?
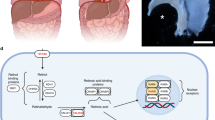
NR2F2 (COUP-TFII), mentioned previously as a potential target of Shh-signalling in neural cells, was also found to be expressed in the murine nonmuscular mesenchyme of the PPFs and is frequently affected by deletions in patients with CDH [37,38,39]. Consistently, Nr2f2-knockout mice develop diaphragmatic hernia [40]. Interestingly, multiple pathways unite upon this essential transcription factor (Supplemental Figure 1), which was found to colocalize with WT1, likewise expressed in PPF connective tissue [37] and to interact with GATA4 and FOG2 [41]. We are unable to provide possible mechanisms of action for all of the CNVs described in impaired diaphragm development. However, a comprehensive list of additional CNVs associated with CDH is presented in Supplemetary Table 1.
Point mutations associated with CDH SNVs
The pinpoint for candidate genes encompassed by CNVs
Besides complex CNVs, also single nucleotide variations (SNVs) are associated with cases of CDH. Some of these mutation sites correspond to regions where CNVs have already been detected thus highlighting significant genes within the deleted or duplicated loci, e.g., NR2F2 [42] or GATA4 [43] (Suppl. Table 2). Similar to GATA4, GATA6 is another zinc-finger transcription factor involved particularly in heart and lung organogenesis [44, 45] and interestingly, CDH cases exhibiting GATA6 variants were also reported [46, 47]. Notably, GATA6 expression is restricted to the mesenchymal part of the PPFs [48]. The activity levels of GATA4 and GATA6 are both modulated by the zinc-finger transcription factor FOG2 (also known as ZFPM2) thus activating or repressing transcriptional activity. Hence, mutations affecting FOG2 could also interfere with the diaphragm, heart, and lung development. At E13.5, FOG2 and GATA4 were found to be expressed in nuclei in cells of the PPF. Conversely at E16.5, GATA4 and FOG2 were also determined in the muscularized diaphragm though only FOG2 switched localisation to cytosolic [37]. To our knowledge, Ackerman et al. were the first to discover FOG2 variants in association with CDH [49]. In their experiments, Fog2+/- mice displayed pulmonary dysplasia and the absence of an accessory right lobe along with an intact but muscular posterior and peripheral diaphragm [49]. Furthermore, Fog2+/- mice exhibited severely downregulated HGF expression in the PPFs. How FOG2 might influence the developing diaphragm is not yet fully understood. However, the authors concluded, that impaired migration of muscle cell progenitors due to defective HGF/c-MET signalling led to the non-muscularized diaphragm and hypothesised HGF expression might be directly regulated by FOG2. Also, FOG2 is thought to act as a corepressor of NR2F2 [50]. Consistently, SNVs in MET were found in cases of CDH [23, 51] supporting the concept of defective migration as a cause for high diaphragmatic vulnerability.
The role of vitamin A homoeostasis and signalling pathways
There is scientific consensus about the importance of vitamin A and its derivatives in embryonic lung development [52, 53]. Tissue-specific retinaldehyde dehydrogenases (RALDHs) generate retinoic acids (RAs) in two different isoforms, namely all-trans retinoic acid and 9-cis retinoic acid both binding to different intracellular receptors (RAR or RXR), which then form heterodimers and translocate to the nucleus, where they act as transcription factors by binding to specific retinoic acid response elements (RAREs, RXREs). During alveologenesis, RALDH2 (ALDH1A2) and ALDH1 specifically expressed in lung tissue are of significant importance for generating RAs [54], which are thought to perpetuate mesodermal proliferation, to induce FGF10 and to affect shh-signalling during the period of initial budding and branching morphogenesis [52, 55, 56]. Also, retinoic acids act upstream of homeobox transcription factors like HOXB4 to promote lung differentiation [57]. Interestingly, there are mutated variants of HOXB4 associated with CDH [23]. Supplemental Figure 1 provides an overview on vitamin A’s metabolism and target genes in lung development. The observation, that vitamin A deficiency during rat dams’ pregnancy did not only impede lung development but did also cause diaphragm defects in the pups led to the hypothesis that vitamin A signalling would be involved in its morphogenesis [58, 59]. Indeed, Aldh1a2, retinol-binding protein Rbp, and Rarβ were found to be strongly expressed in the PPFs of rat pups [60,61,62] and generated Rar-knockout strains exhibiting diaphragmatic defects additional to pulmonary hypoplasia further highlight the importance of vitamin A in diaphragm development [61]. Conversely, Clugston et al. evaluated the presence of Rarα, Rarγ and Rxrα but did not determine Rarβ transcripts in the developing murine diaphragm [63]. Subsequently performed studies on human newborns revealed a reduction of retinol and RBP in the chord blood by up to 50% in CDH cases [64]. In consistence with the emerging concept of vitamin A-dependent diaphragm development, it was shown NR2F2 (COUP-TFII) transcription factors could act as repressors of and in turn be regulated by retinoic acid signalling [65, 66] and by this, a possible mechanism in CDH pathogenesis was revealed. Supporting the concept of essential vitamin A signalling in diaphragm development, both point mutations in and CNVs of genes involved in vitamin A metabolism and signalling were described affecting several levels of retinoid signalling. Starting at the top of the signalling cascade, SNVs in STRA6 (encoding a membrane receptor involved in the uptake of vitamin A) and a CNV covering STRA6 potentially compromising vitamin A uptake in affected cells were discovered [67, 68]. Furthermore, ALDH1A2 was found to be affected by duplications on chromosome 9 in CDH cases [69, 70]. Interestingly, one of those duplications also covered the retinoid acid receptors RARA and RXRA, the terminal endpoints of retinoid signalling [69]. Given the numerous mutations in members of the retinoid signalling pathway discovered in CDH patients (Supplementary Table 1 and 2), we provide evidence for its significant contributions to diaphragm development and CDH pathogenesis.
Discussion
We comprehensively summarise point mutations and copy number variations described in the literature associated with CDH. Both have contributed equally to the identification of CDH candidate genes and are nowadays indispensable in its deep diagnosis. However, declaring detected genes explicitly causative would be a bold statement with regards to the complexity of genetics and organogenesis during embryonic development. Because deleted or duplicated chromosomal loci often carry many different genes, a disease-causing genotype-phenotype association is a difficult venture. Also, the deletion or duplication of promotor regions or regulatory units could possibly escape our notice. Moreover, the fact that in some cases unaffected relatives harbour the same genetic anomaly further underlines the complexity of CDH pathogenesis. Nevertheless, our summary of genetic findings associated with CDH offers the possibility for clinicians and geneticist to classify rare mutations in affected infants as potentially relevant, and subsequently to screen parents for the respective genetic variant and offer genetic counselling. By doing so, future pregnancies rated high-risk upon the presence of certain mutations could be closely monitored in order to improve the outcome of the newborn. The development of diaphragmatic hernias can be considered the endpoint of a single (rare), dual, or even multiple hit event in multiple tissues or the interaction between cells of different ancestry. We consider mutational events in the pathogenesis of CDH as being of predisposing character as it remains difficult to make single specific events solely responsible for the development of diaphragmatic defects. Mutated genes can be assigned to multiple signalling pathways (e.g., retinoid, hedgehog, wnt), play a role in various areas of cellular homoeostasis or are crucial during embryonic development. The majority resembles transcription factors of which some had been exhaustively investigated and their presence in the premature murine diaphragm or pleuroperitoneal folds have been determined. However, we think more transcription factors and proteins could be found in the premature diaphragm and further extend the spectrum of CDH candidate genes and possible pathogenic backgrounds. There seems to be more independent factors explaining why some individuals with certain genetic anomalies do develop CDH and others do not. Likewise, genetics alone could hardly explain the local restriction of CDH defects. Furthermore, there is evidence for essential vitamin A signalling in the developing primitive lungs and diaphragm. Disruption of the retinoid signalling pathway would therefore be able to constitute a basis for an in vivo applicable dual hit hypothesis, suggesting that lung hypoplasia could in part occur independently of a diaphragmatic defect or even precede it. In the literature, possible pathways by which vitamin A could impede diaphragm development are poorly discussed. Hence, its contribution to the pathogenicity of CDH remains largely unknown. By demonstrating RA-inducible NR2F2 activity, at least one possible mechanism of RA-regulated CDH development has been revealed. Further research needs to be done to verify if there are interactions between retinoic acid and other transcription factors known to be crucial for diaphragm development.
Data availability
All data generated or analysed during this study are included in this published article [and its supplementary information files].
References
Doyle NM, Lally KP, eds. The CDH Study Group and advances in the clinical care of the patient with congenital diaphragmatic hernia. Seminars in perinatology; 2004: Elsevier.
Tovar JA. Congenital diaphragmatic hernia. Orphanet J Rare Dis. 2012;7:1–15.
Snoek KG, Reiss IK, Greenough A, Capolupo I, Urlesberger B, Wessel L, et al. Standardized postnatal management of infants with congenital diaphragmatic hernia in Europe: the CDH EURO Consortium Consensus-2015 update. Neonatology 2016;110:66–74.
Montalva L, Raffler G, Riccio A, Lauriti G, Zani A. Neurodevelopmental impairment in children with congenital diaphragmatic hernia: not an uncommon complication for survivors. J Pediatr Surg. 2019;55:625–34.
Ameis D, Khoshgoo N, Keijzer R, eds. Abnormal lung development in congenital diaphragmatic hernia. Seminars in pediatric surgery; 2017: Elsevier.
Cannata G, Caporilli C, Grassi F, Perrone S, Esposito S. Management of congenital diaphragmatic hernia (CDH): role of molecular genetics. Int J Mol Sci. 2021;22:6353.
Yu L, Hernan RR, Wynn J, Chung WK, eds. The influence of genetics in congenital diaphragmatic hernia. Seminars in perinatology; 2020: Elsevier.
Iritani I. Experimental study on embryogenesis of congenital diaphragmatic hernia. Anat Embryol. 1984;169:133–9.
Allan DW, Greer JJ. Embryogenesis of the phrenic nerve and diaphragm in the fetal rat. J Comp Neurol. 1997;382:459–68.
Ionita-Laza I, Rogers AJ, Lange C, Raby BA, Lee C. Genetic association analysis of copy-number variation (CNV) in human disease pathogenesis. Genomics 2009;93:22–6.
McCarroll SA, Altshuler DM. Copy-number variation and association studies of human disease. Nat Genet. 2007;39:S37.
Weischenfeldt J, Symmons O, Spitz F, Korbel JO. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat Rev Genet. 2013;14:125.
Pober BR, editor. Overview of epidemiology, genetics, birth defects, and chromosome abnormalities associated with CDH. American Journal of Medical Genetics Part C: Seminars in Medical Genetics; 2007: Wiley Online Library.
Zhu Q, High FA, Zhang C, Cerveira E, Russell MK, Longoni M, et al. Systematic analysis of copy number variation associated with congenital diaphragmatic hernia. Proc Natl Acad Sci USA. 2018;115:5247–52.
Lints T, Hartley L, Parsons L, Harvey R. Mesoderm‐specific expression of the divergent homeobox gene Hlx during murine embryogenesis. Developmental Dyn: Off Publ Am Assoc Anatomists. 1996;205:457–70.
Slavotinek A, Moshrefi A, Lopez Jiminez N, Chao R, Mendell A, Shaw G, et al. Sequence variants in the HLX gene at chromosome 1q41‐1q42 in patients with diaphragmatic hernia. Clin Genet. 2009;75:429–39.
Ma Y, Erkner A, Gong R, Yao S, Taipale J, Basler K, et al. Hedgehog-mediated patterning of the mammalian embryo requires transporter-like function of dispatched. Cell 2002;111:63–75.
Kantarci S, Ackerman KG, Russell MK, Longoni M, Sougnez C, Noonan KM, et al. Characterization of the chromosome 1q41q42. 12 region, and the candidate gene DISP1, in patients with CDH. Am J Med Genet Part A. 2010;152:2493–504.
Liem KF, He M, Ocbina PJR, Anderson KV. Mouse Kif7/Costal2 is a cilia-associated protein that regulates Sonic hedgehog signaling. Proc Natl Acad Sci. 2009;106:13377–82.
Krishnan V, Pereira FA, Qiu Y, Chen C-H, Beachy PA, Tsai SY, et al. Mediation of Sonic hedgehog-induced expression of COUP-TFII by a protein phosphatase. Science 1997;278:1947–50.
Pepicelli CV, Lewis PM, McMahon AP. Sonic hedgehog regulates branching morphogenesis in the mammalian lung. Curr Biol. 1998;8:1083–6.
Jordan VK, Beck TF, Hernandez-Garcia A, Kundert PN, Kim B-J, Jhangiani SN, et al. The role of FREM2 and FRAS1 in the development of congenital diaphragmatic hernia. Hum Mol Genet. 2018;27:2064–75.
Longoni M, High FA, Russell MK, Kashani A, Tracy AA, Coletti CM, et al. Molecular pathogenesis of congenital diaphragmatic hernia revealed by exome sequencing, developmental data, and bioinformatics. Proc Natl Acad Sci. 2014;111:12450–5.
Paris ND, Coles GL, Ackerman KG. Wt1 and β-catenin cooperatively regulate diaphragm development in the mouse. Developmental Biol. 2015;407:40–56.
Sato A, Yamamoto H, Sakane H, Koyama H, Kikuchi A. Wnt5a regulates distinct signalling pathways by binding to Frizzled2. The. EMBO J. 2010;29:41–54.
Wat MJ, Veenma D, Hogue J, Holder AM, Yu Z, Wat JJ, et al. Genomic alterations that contribute to the development of isolated and non-isolated congenital diaphragmatic hernia. J Med Genet. 2011;48:299–307.
Scott D, Cooper M, Stankiewicz P, Patel A, Potocki L, Cheung S. Congenital diaphragmatic hernia in WAGR syndrome. Am J Med Genet Part A. 2005;134:430–3.
Yu L, Wynn J, Ma L, Guha S, Mychaliska GB, Crombleholme TM, et al. De novo copy number variants are associated with congenital diaphragmatic hernia. J Med Genet. 2012;49:650–9.
Keitges EA, Pasion R, Burnside RD, Mason C, Gonzalez‐Ruiz A, Dunn T, et al. Prenatal diagnosis of two fetuses with deletions of 8p23. 1, critical region for congenital diaphragmatic hernia and heart defects. Am J Med Genet Part A. 2013;161:1755–8.
Rajagopal SK, Ma Q, Obler D, Shen J, Manichaikul A, Tomita-Mitchell A, et al. Spectrum of heart disease associated with murine and human GATA4 mutation. J Mol Cell Cardiol. 2007;43:677–85.
Reamon-Buettner S, Borlak J. GATA4 zinc finger mutations as a molecular rationale for septation defects of the human heart. J Med Genet. 2005;42:e32-e.
Lentjes MH, Niessen HE, Akiyama Y, De Bruine AP, Melotte V, Van Engeland M. The emerging role of GATA transcription factors in development and disease. Expert Rev Mol Med. 2016;18:e3.
Jay PY, Bielinska M, Erlich JM, Mannisto S, Pu WT, Heikinheimo M, et al. Impaired mesenchymal cell function in Gata4 mutant mice leads to diaphragmatic hernias and primary lung defects. Developmental Biol. 2007;301:602–14.
Merrell AJ, Ellis BJ, Fox ZD, Lawson JA, Weiss JA, Kardon G. Muscle connective tissue controls development of the diaphragm and is a source of congenital diaphragmatic hernias. Nat Genet. 2015;47:496.
Wat MJ, Shchelochkov OA, Holder AM, Breman AM, Dagli A, Bacino C, et al. Chromosome 8p23. 1 deletions as a cause of complex congenital heart defects and diaphragmatic hernia. Am J Med Genet Part A. 2009;149:1661–77.
Futaki S, Hayashi Y, Emoto T, Weber CN, Sekiguchi K. Sox7 plays crucial roles in parietal endoderm differentiation in F9 embryonal carcinoma cells through regulating Gata-4 and Gata-6 expression. Mol Cell Biol. 2004;24:10492–503.
Clugston RD, Zhang W, Greer JJ. Gene expression in the developing diaphragm: significance for congenital diaphragmatic hernia. Am J Physiol-Lung Cell Mol Physiol. 2008;294:L665–L75.
Brady P, DeKoninck P, Fryns J-P, Devriendt K, Deprest J, Vermeesch J. Identification of dosage‐sensitive genes in fetuses referred with severe isolated congenital diaphragmatic hernia. Prenat diagnosis. 2013;33:1283–92.
Klaassens M, van Dooren M, Eussen H, Douben H, Den Dekker A, Lee C, et al. Congenital diaphragmatic hernia and chromosome 15q26: determination of a candidate region by use of fluorescent in situ hybridization and array-based comparative genomic hybridization. Am J Hum Genet. 2005;76:877–82.
You L-R, Takamoto N, Yu C-T, Tanaka T, Kodama T, DeMayo FJ, et al. Mouse lacking COUP-TFII as an animal model of Bochdalek-type congenital diaphragmatic hernia. Proc Natl Acad Sci. 2005;102:16351–6.
Brady PD, Srisupundit K, Devriendt K, Fryns J-P, Deprest JA, Vermeesch JR. Recent developments in the genetic factors underlying congenital diaphragmatic hernia. Fetal diagnosis Ther. 2011;29:25–39.
High FA, Bhayani P, Wilson JM, Bult CJ, Donahoe PK, Longoni M. De novo frameshift mutation in COUP‐TFII (NR2F2) in human congenital diaphragmatic hernia. Am J Med Genet Part A. 2016;170:2457–61.
Yu L, Wynn J, Cheung YH, Shen Y, Mychaliska GB, Crombleholme TM, et al. Variants in GATA4 are a rare cause of familial and sporadic congenital diaphragmatic hernia. Hum Genet. 2013;132:285–92.
Keijzer R, van Tuyl M, Meijers C, Post M, Tibboel D, Grosveld F, et al. The transcription factor GATA6 is essential for branching morphogenesis and epithelial cell differentiation during fetal pulmonary development. Development 2001;128:503–11.
Xin M, Davis CA, Molkentin JD, Lien C-L, Duncan SA, Richardson JA, et al. A threshold of GATA4 and GATA6 expression is required for cardiovascular development. Proceedings of the National Academy of Sciences. 2006;103:11189–94.
Allen HL, Flanagan SE, Shaw-Smith C, De Franco E, Akerman I, Caswell R, et al. GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat Genet. 2012;44:20.
Gaisl OC, Konrad D, Steindl K, Lang-Muritano M, eds. Novel Gata6-Mutation in a Boy with Neonatal Diabetes and Diaphragmatic Hernia. 57th Annual ESPE; 2018: European Society for Paediatric Endocrinology.
Takahashi T, Friedmacher F, Zimmer J, Puri P. Gata-6 expression is decreased in diaphragmatic and pulmonary mesenchyme of fetal rats with nitrofen-induced congenital diaphragmatic hernia. Pediatr Surg Int. 2018;34:315–21.
Ackerman KG, Herron BJ, Vargas SO, Huang H, Tevosian SG, Kochilas L, et al. Fog2 is required for normal diaphragm and lung development in mice and humans. PLoS Genet. 2005;1:e10.
Huggins GS, Bacani CJ, Boltax J, Aikawa R, Leiden JM. Friend of GATA 2 physically interacts with chicken ovalbumin upstream promoter-TF2 (COUP-TF2) and COUP-TF3 and represses COUP-TF2-dependent activation of the atrial natriuretic factor promoter. J Biol Chem. 2001;276:28029–36.
Beck TF, Campeau PM, Jhangiani SN, Gambin T, Li AH, Abo‐Zahrah R, et al. FBN1 contributing to familial congenital diaphragmatic hernia. Am J Med Genet Part A. 2015;167:831–6.
Maden M. 7 Retinoids in Lung Development and Regeneration. Curr Top developmental Biol. 2004;61:154–91.
Malpel S, Mendelsohn C, Cardoso WV. Regulation of retinoic acid signaling during lung morphogenesis. Development 2000;127:3057–67.
Hind M, Corcoran J, Maden M. Alveolar proliferation, retinoid synthesizing enzymes, and endogenous retinoids in the postnatal mouse lung: different roles for Aldh-1 and Raldh-2. Am J respiratory cell Mol Biol. 2002;26:67–73.
Desai TJ, Malpel S, Flentke GR, Smith SM, Cardoso WV. Retinoic acid selectively regulates Fgf10 expression and maintains cell identity in the prospective lung field of the developing foregut. Developmental Biol. 2004;273:402–15.
Rankin SA, Han L, McCracken KW, Kenny AP, Anglin CT, Grigg EA, et al. A retinoic acid-hedgehog cascade coordinates mesoderm-inducing signals and endoderm competence during lung specification. Cell Rep. 2016;16:66–78.
Volpe MV, Wang KTW, Nielsen HC, Chinoy MR. Unique spatial and cellular expression patterns of Hoxa5, Hoxb4, and Hoxb6 proteins in normal developing murine lung are modified in pulmonary hypoplasia. Birth Defects Res Part A: Clin Mol Teratol. 2008;82:571–84.
Andersen D. Incidence of congenital diaphragmatic hernia in the young of rats bred on a diet deficient in vitamin A. Am J Dis Child. 1941;62:888–9.
Wilson JG, Roth CB, Warkany J. An analysis of the syndrome of malformations induced by maternal vitamin A deficiency. Effects of restoration of vitamin A at various times during gestation. Am J Anat. 1953;92:189–217.
Båvik C, Ward SJ, Ong DE. Identification of a mechanism to localize generation of retinoic acid in rat embryos. Mechanisms Dev. 1997;69:155–67.
Mendelsohn C, Lohnes D, Décimo D, Lufkin T, LeMeur M, Chambon P, et al. Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development 1994;120:2749–71.
Mey J, Babiuk RP, Clugston R, Zhang W, Greer JJ. Retinal dehydrogenase-2 is inhibited by compounds that induce congenital diaphragmatic hernias in rodents. Am J Pathol. 2003;162:673–9.
Clugston RD, Zhang W, Álvarez S, De Lera AR, Greer JJ. Understanding abnormal retinoid signaling as a causative mechanism in congenital diaphragmatic hernia. Am J respiratory cell Mol Biol. 2010;42:276–85.
Major D, Cadenas M, Fournier L, Leclerc S, Lefebvre M, Cloutier R. Retinol status of newborn infants with congenital diaphragmatic hernia. Pediatr Surg Int. 1998;13:547–9.
Cooney AJ, Tsai SY, O’Malley BW, Tsai M. Chicken ovalbumin upstream promoter transcription factor (COUP-TF) dimers bind to different GGTCA response elements, allowing COUP-TF to repress hormonal induction of the vitamin D3, thyroid hormone, and retinoic acid receptors. Mol Cell Biol. 1992;12:4153–63.
Qiu Y, Krishnan V, Pereira F, Tsai S, Tsai M-J. Chicken ovalbumin upstream promoter-transcription factors and their regulation. J steroid Biochem Mol Biol. 1996;56:81–5.
Pasutto F, Sticht H, Hammersen G, Gillessen-Kaesbach G, FitzPatrick DR, Nürnberg G, et al. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am J Hum Genet. 2007;80:550–60.
Van Esch H, Backx L, Pijkels E, Fryns J-P. Congenital diaphragmatic hernia is part of the new 15q24 microdeletion syndrome. Eur J Med Genet. 2009;52:153–6.
Henriques-Coelho T, Oliva-Teles N, Fonseca-Silva ML, Tibboel D, Guimarães H, Correia-Pinto J. Congenital diaphragmatic hernia in a patient with tetrasomy 9p. J Pediatr Surg. 2005;40:e29–e31.
Steiner MB, Vengoechea J, Collins RT. Duplication of the ALDH1A2 gene in association with pentalogy of Cantrell: a case report. J Med case Rep. 2013;7:287.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schreiner, Y., Schaible, T. & Rafat, N. Genetics of diaphragmatic hernia. Eur J Hum Genet 29, 1729–1733 (2021). https://doi.org/10.1038/s41431-021-00972-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-021-00972-0
This article is cited by
-
Genomics elucidates both common and rare disease aetiology
European Journal of Human Genetics (2021)
