
Abstract
Germline structural variants (SVs) are challenging to resolve by conventional genetic testing assays. Long-read sequencing has improved the global characterization of SVs, but its sensitivity at cancer susceptibility loci has not been reported. Nanopore long-read genome sequencing was performed for nineteen individuals with pathogenic copy number alterations in BRCA1, BRCA2, CHEK2 and PALB2 identified by prior clinical testing. Fourteen variants, which spanned single exons to whole genes and included a tandem duplication, were accurately represented. Defining the precise breakpoints of SVs in BRCA1 and CHEK2 revealed unforeseen allelic heterogeneity and informed the mechanisms underlying the formation of recurrent deletions. Integrating read-based and statistical phasing further helped define extended haplotypes associated with founder alleles. Long-read sequencing is a sensitive method for characterizing private, recurrent and founder SVs underlying breast cancer susceptibility. Our findings demonstrate the potential for nanopore sequencing as a powerful genetic testing assay in the hereditary cancer setting.
Similar content being viewed by others
Introduction
Breast cancer is the most common cancer in females with an estimated 2.3 million new diagnoses worldwide in 2020 [1]. Rare variants in genes associated with high-penetrance cancer predisposition syndromes confer a strong genetic susceptibility in around 5–10% of cases depending on ascertainment criteria. BRCA1, BRCA2, and the moderate-penetrance genes CHEK2 and PALB2 have also been associated with increased risks for male breast cancer, a rare disease in which 18% of cases may be related to clinically actionable germline variants [2, 3]. Identifying carriers for moderate- to high-penetrance variants can improve clinical outcomes by informing disease risk and prognosis, and guide recommendations for prophylactic intervention, cancer screening and therapy. However, many individuals who undergo genetic testing based on a strong personal or family history of breast and other syndrome-related cancers receive uninformative results [4].
Short-read sequencing (SRS) is the most common technology used in clinical laboratories for genetic testing due to its high throughput, high analytic validity, and low relative cost. Despite these advantages, one in seven pathogenic germline variants are challenging to detect using SRS [5]. Long-read sequencing (LRS) has shown potential for improving rates of molecular diagnosis by more accurately identifying structural variants (SVs) and repeat expansions, resolving complex rearrangements, and informing phase of candidate variants [6]. Although the clinical utility of LRS has been described in the diagnosis of various genetic syndromes, the sensitivity of long-read technologies for characterizing structural variation at loci commonly tested in the clinical setting is not well-established.
Here, we assessed the accuracy of nanopore long-read genome sequencing (GS) for characterizing pathogenic germline SVs in four breast cancer susceptibility genes. Expanding upon results from clinical testing, precise breakpoints could be defined at nucleotide resolution, revealing uncharacterized allelic heterogeneity at the loci of recurrent and founder variants. Our findings may inform the future implementation of LRS as an alternative to standard clinical assays.
Materials and methods
This study was approved by the University of British Columbia Clinical Research Ethics Board (H19-01594). All participants provided written informed consent. PCR-free genome libraries were prepared from DNA isolated from peripheral blood lymphocytes and sequenced on the Oxford Nanopore Technologies PromethION. Single nucleotide variant (SNV) and small insertion and deletion (indel) calling and phasing were performed using an established pipeline [7]. SVs were manually reviewed in IGV. SV breakpoints were defined by local assembly-derived contigs where possible, and reported according to HGVS sequence variant nomenclature. Haplotype inference was performed using integrated read- and population-based phasing [8]. Please refer to the Supplementary Materials for detailed methods.
Results
GS was performed for 19 individuals from 18 families with pathogenic deletions and duplications in BRCA1, BRCA2, CHEK2 or PALB2 identified by prior clinical testing (Supplementary Table S1). Individuals were referred for index or carrier testing on the basis of a suspected inherited predisposition to breast cancer or known familial variant, respectively. Sequencing was performed to a median coverage of 21.6X (13.1–36.5X), achieving a median read N50 of 14.5 kb (6.57–23.5 kb) (Supplementary Table S2).
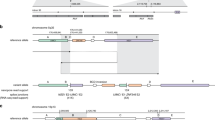
Variants in all 19 carriers, representing 14 distinct SVs ranging in size from 510 bp to 108 kb, were detected by nanopore sequencing (Table 1). While some variants were not identified by agnostic SV calling, all known SVs were supported by at least three reads (Supplementary Table S2). The precise breakpoints were refined in all but one case with low coverage and low complexity sequence at one breakpoint. Previously unknown allelic heterogeneity was revealed at the locus of BRCA1: three deletions spanning BRCA1 exons 1–2 were characterized by intragenic breakpoints within 3.7 kb in intron 2 (Fig. 1). Telomeric breakpoints in NBR2 and LOC101929767 (ΨBRCA1), a partial BRCA1 pseudogene, were associated with deletions of 6.6 kb and 36–37 kb, respectively. These findings were consistent with previous observations of recurrent deletions between BRCA1 and adjacent loci [9].

Read alignments and contigs derived by local assembly of variant supporting reads at the BRCA1 locus. Telomeric breakpoints within intron 1 of the BRCA1 pseudogene were associated with deletions of 37,470 bp (P1) and 36,056 bp (P2), and breakpoints within NBR2 intron 1 were associated with a deletion of 6649 bp (P3). For clarity, mismatch bases and indels <50 bp are not shown.
Among founder populations, specific genetic variants make a considerable contribution to disease susceptibility. To explore the potential for nanopore GS to characterize founder haplotypes, we integrated read-based and statistical phasing using a reference haplotype panel from 2504 individuals sequenced as part of the 1000 Genomes Project Phase 3 [8]. For genomes with at least 20X average coverage (n = 13), read information alone allowed phasing for 77–92% of heterozygous SNVs, and longer reads were associated with larger haplotype blocks (Spearman correlation 0.78; Supplementary Fig. S1) [7]. Long reads spanning breakpoints of the British BRCA1 founder duplication (ins6kbEx13) confirmed a 6126 bp tandem duplication in three unrelated individuals (Supplementary Fig. S2) [10]. Analysis of SNVs extending beyond the boundaries of the BRCA1 ins6kbEx13 founder variant further defined a core 1.08 Mb haplotype shared between carriers.
Five individuals had deletions of CHEK2 exons 9–10, characteristic of a 5395 bp deletion (del5395) estimated to account for 1% of breast cancers in Poland [11, 12]. LRS confirmed the CHEK2 del5395 founder variant in three individuals; however, two related individuals had a larger 6188 bp deletion (del6188) with breakpoints in two Alu short interspersed nuclear elements with 71% sequence identity (Fig. 2 and Supplementary Fig. S2). Two base pair regions of microhomology at the breakpoints of the former suggest the del5395 and del6188 variants originated through distinct microhomology- and recombination-mediated mechanisms of formation, respectively. The CHEK2 del5395 variant was associated with a core 1.26 Mb haplotype characterized by a rare SNV in cis located 100 kb upstream and specific to carriers of the del5395 founder variant (Supplementary Fig. S3). Together, these findings suggested that the del5395 and del6188 variants were unlikely to have arisen from the same subpopulation.

Read alignments and contigs derived by local assembly of variant supporting reads at the CHEK2 locus. Reads are colored in blue and pink by haplotype inferred from integrated read-based and reference-guided phasing. Reads that could not be assigned to a haplotype are colored in gray. Local haploid assemblies were generated from reads supporting each variant to refine the breakpoints of the CHEK2 del5395 European founder variant (P11) and the CHEK2 del6188 variant (P14) in representative carriers. SINEs short interspersed nuclear elements. For clarity, mismatch bases and indels <50 bp are not shown.
Discussion
Advances in sequencing technologies have revealed a greater spectrum of heritable variation underlying human diversity and disease. Given the variable expressivity and genetic heterogeneity of breast cancer predisposition syndromes, multigene panel SRS has become widespread practice to identify families with an increased risk for disease. However, limitations of standard clinical assays for identifying complex genetic changes may underestimate the contribution of SVs to cancer susceptibility. Nanopore GS resolved 14 distinct copy number variants in high- and moderate-penetrance genes across 19 individuals with known breast cancer susceptibility. Our findings reveal unexpected allelic heterogeneity at the locus of CHEK2, and demonstrate the potential for LRS to characterize haplotype-resolved structural variation in personal genomes.
Characterizing the molecular heterogeneity of pathogenic variants in cancer susceptibility genes may inform estimates of individual cancer risk. Deletions of BRCA1 exons 1–2 account for 10–15% of pathogenic copy number variants in BRCA1 [13]. Consistent with previous reports, we identified recurrent deletions between 6.6 and 37 kb in three individuals with variable loss of BRCA1 exons 1–2 and the 5’ region upstream. Targeted clinical testing could not reveal the extent of genetic loss. Among five individuals from four families with deletions of CHEK2 exons 9–10, two related individuals were found to carry a 6188 bp deletion distinct from the del5395 Eastern European founder variant. Resolving the precise breakpoints of SVs may thus inform their molecular origins and natural history, and allow the development of customized confirmation assays for rapid and accurate carrier screening.
To a greater extent than deletions, the clinical interpretation of duplications remains challenging for SRS. Importantly, determining the location and orientation of duplications can inform the etiology of disease [14]. Nanopore sequencing accurately mapped the breakpoints of a tandem duplication and known founder variant, BRCA1 ins6kbEx13, in three individuals. Despite sufficient read coverage, this variant was not identified by available SV callers, indicating a need for further development of SV detection methods using long reads. LRS has also shown potential to characterize cryptic, copy neutral and complex rearrangements whose clinical or functional significance is uncertain [15, 16]. These variants may remain undetected or unresolved by SRS, suggesting their contribution to cancer susceptibility may be underappreciated.
Using read-based and reference-guided phasing, we defined haplotypes shared between carriers of founder variants and identified rare alleles in cis that are likely to be identical by descent from a common ancestor. For diseases with common genetic aetiologies, chromosome-scale haplotyping may uncover alleles associated with causal variants in silent carriers who would otherwise go undetected [17]. The accuracy of reference-guided phasing depends on the composition and size of reference panels however, and many populations remain underrepresented in current population databases [18]. Therefore, large-scale efforts to characterize genetic variation across subpopulations of diverse genetic ancestries are needed.
The throughput and analytical validity of LRS have improved rapidly in the past several years with advances in Oxford Nanopore Technologies’ nanopore sequencing and Pacific Biosciences single-molecule, real-time sequencing. Recent library preparation and pore chemistries have allowed the sensitivity of SNV and indel calling from nanopore sequencing to exceed 99% in the coding genome [19]. The costs of nanopore GS, around $1500–$2000 CAD per sample, limit its wider clinical application compared to under $500 CAD for current clinical multigene panels. However, library multiplexing and PCR-free enrichment methods, including Cas9-mediated enrichment and computational selection by adaptive sampling, will enable cost-effective targeted nanopore sequencing whose throughput and accuracy could be comparable to multigene panel SRS [6, 20]. LRS thus offers a comprehensive testing strategy that may soon be readily adoptable in local diagnostic laboratories for routine testing of hereditary cancer susceptibility.
Data availability
Raw sequencing data has been deposited in the European Genome-Phenome Archive as part of study EGAS00001005872. All scripts included in this work are available upon request.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Pritzlaff M, Summerour P, McFarland R, Li S, Reineke P, Dolinsky JS, et al. Male breast cancer in a multi-gene panel testing cohort: insights and unexpected results. Breast Cancer Res Treat. 2017;161:575–86.
Chamseddine RS, Wang J, Yin K, Singh P, Zhou J, Braun D, et al. Abstract PS8-40: Penetrance of male breast cancer susceptibility genes: a systematic review. Cancer Res. 2021;81:PS8–40.
LaDuca H, Stuenkel AJ, Dolinsky JS, Keiles S, Tandy S, Pesaran T, et al. Utilization of multigene panels in hereditary cancer predisposition testing: analysis of more than 2,000 patients. Genet Med. 2014;16:830–7.
Lincoln SE, Hambuch T, Zook JM, Bristow SL, Hatchell K, Truty R, et al. One in seven pathogenic variants can be challenging to detect by NGS: an analysis of 450,000 patients with implications for clinical sensitivity and genetic test implementation. Genet Med. 2021;23:1673–80.
Miller DE, Sulovari A, Wang T, Loucks H, Hoekzema K, Munson KM, et al. Targeted long-read sequencing identifies missing disease-causing variation. Am J Hum Genet. 2021;108:1436–49.
Shafin K, Pesout T, Lorig-Roach R, Haukness M, Olsen HE, Bosworth C, et al. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat Biotechnol. 2020;38:1044–53.
Delaneau O, Zagury J-F, Robinson MR, Marchini JL, Dermitzakis ET. Accurate, scalable and integrative haplotype estimation. Nat Commun. 2019;10:5436.
Puget N, Gad S, Perrin-Vidoz L, Sinilnikova OM, Stoppa-Lyonnet D, Lenoir GM, et al. Distinct BRCA1 rearrangements involving the BRCA1 pseudogene suggest the existence of a recombination hot spot. Am J Hum Genet. 2002;70:858–65.
Puget N, Sinilnikova OM, Stoppa-Lyonnet D, Audoynaud C, Pages S, Lynch HT, et al. An Alu-mediated 6-kb duplication in the BRCA1 gene: a new founder mutation? Am J Hum Genet. 1999;64:300–2.
Walsh T, Casadei S, Coats KH, Swisher E, Stray SM, Higgins J, et al. Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA. 2006;295:1379–88.
Cybulski C, Wokołorczyk D, Huzarski T, Byrski T, Gronwald J, Górski B, et al. A deletion in CHEK2 of 5,395 bp predisposes to breast cancer in Poland. Breast Cancer Res Treat. 2006;102:119–22.
Caputo SM, Telly D, Briaux A, Sesen J, Ceppi M, Bonnet F, et al. 5′ region large genomic rearrangements in the BRCA1 gene in French families: identification of a tandem triplication and nine distinct deletions with five recurrent breakpoints. Cancers. 2021;13:17.
Newman S, Hermetz KE, Weckselblatt B, Rudd MK. Next-generation sequencing of duplication CNVs reveals that most are tandem and some create fusion genes at breakpoints. Am J Hum Genet. 2015;96:208.
Thibodeau ML, O’Neill K, Dixon K, Reisle C, Mungall KL, Krzywinski M, et al. Improved structural variant interpretation for hereditary cancer susceptibility using long-read sequencing. Genet Med. 2020;22:1892–7.
Walsh T, Casadei S, Munson KM, Eng M, Mandell JB, Gulsuner S, et al. CRISPR–Cas9/long-read sequencing approach to identify cryptic mutations in BRCA1 and other tumour suppressor genes. J Med Genet. 2021;58:850–2.
Luo M, Liu L, Peter I, Zhu J, Scott SA, Zhao G, et al. An Ashkenazi Jewish SMN1 haplotype specific to duplication alleles improves pan-ethnic carrier screening for spinal muscular atrophy. Genet Med. 2014;16:149–56.
Choi Y, Chan AP, Kirkness E, Telenti A, Schork NJ. Comparison of phasing strategies for whole human genomes. PLOS Genet. 2018;14:e1007308.
Accuracy. https://nanoporetech.com/accuracy. Accessed 22 Dec 2022.
Gabrieli T, Sharim H, Fridman D, Arbib N, Michaeli Y, Ebenstein Y. Selective nanopore sequencing of human BRCA1 by Cas9-assisted targeting of chromosome segments (CATCH). Nucleic Acids Res. 2018;46:e87–e87.
Acknowledgements
We wish to gratefully acknowledge the patients and research participants who contributed to this study. This work was supported by the BC Cancer Foundation Neil Macrae Hereditary Cancer Research Fund. KD is supported by Michael Smith Health Research BC. KAS acknowledges funding from the Canada Research Chairs program and Canadian Institutes of Health Research. SJMJ acknowledges funding from the Canada Research Chairs program and the Canadian Foundation for Innovation.
Author information
Authors and Affiliations
Contributions
KD was responsible for data interpretation and writing the manuscript. KD, YS, KO, SC, SB, and WZ were responsible for data analysis. AF, MB, and AS were responsible for recruiting and provided project administration support. KLM, AJM, RM, IB, SY, SS, KAS, and SJMJ provided supervision. MLT, SS, KAS and SJMJ were responsible for study conception and design, and funding acquisition. All authors contributed to the research and critical review of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study was approved by the University of British Columbia Clinical Research Ethics Board (#H19-01594). Study participants were ascertained prospectively and written informed consent was obtained for each participant.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dixon, K., Shen, Y., O’Neill, K. et al. Defining the heterogeneity of unbalanced structural variation underlying breast cancer susceptibility by nanopore genome sequencing. Eur J Hum Genet 31, 602–606 (2023). https://doi.org/10.1038/s41431-023-01284-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-023-01284-1
This article is cited by
-
2023 in the European Journal of Human Genetics
European Journal of Human Genetics (2024)
