
Abstract
Variants of uncertain significance (VUS) are a significant issue for the molecular diagnosis of rare diseases. The publication of episignatures as effective biomarkers of certain Mendelian neurodevelopmental disorders has raised hopes to help classify VUS. However, prediction abilities of most published episignatures have not been independently investigated yet, which is a prerequisite for an informed and rigorous use in a diagnostic setting. We generated DNA methylation data from 101 carriers of (likely) pathogenic variants in ten different genes, 57 VUS carriers, and 25 healthy controls. Combining published episignature information and new validation data with a k-nearest-neighbour classifier within a leave-one-out scheme, we provide unbiased specificity and sensitivity estimates for each of the signatures. Our procedure reached 100% specificity, but the sensitivities unexpectedly spanned a very large spectrum. While ATRX, DNMT3A, KMT2D, and NSD1 signatures displayed a 100% sensitivity, CREBBP-RSTS and one of the CHD8 signatures reached <40% sensitivity on our dataset. Remaining Cornelia de Lange syndrome, KMT2A, KDM5C and CHD7 signatures reached 70–100% sensitivity at best with unstable performances, suffering from heterogeneous methylation profiles among cases and rare discordant samples. Our results call for cautiousness and demonstrate that episignatures do not perform equally well. Some signatures are ready for confident use in a diagnostic setting. Yet, it is imperative to characterise the actual validity perimeter and interpretation of each episignature with the help of larger validation sample sizes and in a broader set of episignatures.

Similar content being viewed by others

Introduction
The efficient etiological diagnosis of neurodevelopmental disorders (NDDs) represents an important matter of public health. However, behind a single denomination, NDDs encompass a wide spectrum of clinical manifestations, arising from a large and heterogeneous set of rare disorders, from monogenic, Mendelian disorders to non-syndromic presentations with variable expression of traits. Providing a timely and accurate molecular diagnosis of monogenic disorders is of utmost importance to patients and their families. A precise and definite molecular diagnosis helps define personalised care for each patient, paves the way to genetic counselling and offers the possibility of prenatal diagnosis. In some cases, the final diagnosis is tediously obtained after years of diagnostic odyssey. Some others remain unsolved.
Large trio-based exome and genome sequencing studies have shown that, beyond environmental factors, genetic factors considerably contribute to the determinism of NDDs. However, genetic causes are extremely heterogeneous. Thousands of genes are now considered to contribute to the genetic etiology of NDDs. In recent years, implementation of exome or genome sequencing in a diagnostic setting has largely contributed to an increase in the rate of patients with definite diagnosis [1]. An important source of improvement is that diagnostic yields result from the rigorous interpretation of (likely) pathogenic (class 4–5) variants according to the ACMG-AMP (American College of Medical Genetics and Genomics—American for Molecular Pathology)—classification [2]. However, the interpretation process is plagued by a large number of variants of uncertain significance (VUS, ACMG-AMP class-3 variants). Finding informative clues to classify VUS as benign or pathogenic is one of the most important tasks in the post-sequencing era of medical genomics.
In the last decade, a new and promising strategy has emerged as an efficient alternative to hard-to-design and time-consuming functional studies to realise this task. The rationale is that a large number of NDD genes are involved in the regulation of gene expression, from transcription factors/regulators to critical players of the dynamic 3D-chromatin organisation and the epigenetic machinery, including histone and DNA methylation regulation [3, 4]. Therefore, the idea is to investigate the pathogenicity of genetic variants through the identification of episignatures, namely vast disruptions in DNA methylation patterns, that are characteristic of affected samples. Over the last 10 years, several research groups have published such episignatures, using Illumina 450 K and more recently 850 K EPIC Infimium Beadchips [5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21]. In practice, episignatures most often result from a two-step approach. First, a genome-wide analysis identifies a set of CpG positions that are differentially methylated between patients affected by a given condition due to a pathogenic variant in a specific set of genes, unique gene or even a specific functional domain, and unaffected controls. Then, a small subset of these positions are combined within a supervised classifier as the final episignature. Support Vector Machines (SVM) are the most common machine-learning approach adopted at this stage in the literature, but any supervised classifier allowing for high-dimensional datasets is possible. Resulting predictions can be used in combination with other arguments from the ACMG-AMP variants interpretation recommendations to discriminate whether a VUS is eventually pathogenic or not.
Several research groups have repeatedly provided evidence of the effectiveness of this concept from a general point of view [18, 22,23,24,25]. More than 50 episignatures have been reported in the literature so far, in the context of more than 60 syndromes [11, 20]. However, for most episignatures, the robustness, reproducibility, and actual sensitivity still need to be assessed on independent datasets, for the following reasons. Firstly, DNA methylation datasets are a perfect example of what statisticians call “ultra-high-dimensional datasets” because the number of CpG positions, here 450 K/800 K, is much larger than the sample size, with sometimes no more than 5 patients [11]. Combining this high-dimensional curse with technical or biological biases, episignatures are prone to overfitting, namely that both the selection step and predictive model might over adjust to the specificities and randomness of the discovery set but will not generalise well to other datasets, with new individuals, generated with different platforms or pipelines. As a result, there is a crucial need to validate the reproducibility of episignature position sets on independent datasets, before we can generalise their use in diagnostic setting. Secondly, because the overall methodology is still varying across the literature [26], an independent evaluation of episignature diagnostic performances (sensitivity and specificity) requires a single neutral and unified framework in which all signatures would be put on the same level for comparison.
With a focus on ten neurodevelopmental disorders, our objective was two-fold. Primarily, we aimed to validate the ability of the 16 corresponding episignatures in the literature to discriminate cases from controls on an independent validation dataset. This ability was quantified through an unbiased assessment of the diagnostic accuracy of corresponding episignatures, in terms of specificity, inter-syndrome specificity and sensitivity. Secondly, we applied our prediction strategy to the classification of VUS and describe the practical challenges encountered. For these purposes, we generated an independent validation and testing set of a target of ten new carrier samples of each tested episignature along with aged and sex-matched controls as well as VUS carriers. Because the actual classification algorithms as well as most of the raw data that would be required to replicate the training steps not always openly-shared, we obtained accurate sensitivity and specificity estimates from our validation dataset by embedding a multiple class k-Nearest Neighbour classification algorithm (kNN) into a leave-one out scheme to estimate the predictive abilities of each published list of probes and then apply it without the leave-one out strategy to VUS samples.
Methods
Sample collection
We leveraged a nation-wide collaborative effort to gather DNA samples isolated from fresh blood of probands harbouring likely pathogenic or pathogenic (LP/P) variants in a list of twelve genes spanning ten neuro-developmental disorders: ATRX (Alpha-thalassemia/mental retardation syndrome, MIM#301040), CHD7 (CHARGE syndrome, MIM#214800), CHD8 (Autism, Susceptibility to, 18 (AUTS18), MIM#615032), CREBBP (Rubinstein-Taybi syndrome 1 (RSTS), MIM#180849), DNMT3A (Tatton-Brown-Rahman syndrome (TBRS), MIM#615879), KDM5C (Intellectual developmental disorder, X-linked syndromic, Claes-Jensen type (MRXSCJ), MIM#300534), KMT2D (Kabuki syndrome 1 MIM# 147920), KMT2A (Wiedemann-Steiner syndrome, MIM#605130), NIPBL/SMC1A/SMC3 (Cornelia de Lange syndrome 1–3 (CdL), MIM#122470,MIM#300590, and MIM#610759), NSD1 (Sotos syndrome, MIM# 117550), as described in Table 1. All these variants were classified as class 4 or 5 variants according to ACMG-AMP interpretation guidelines by experienced geneticists from a network of reference centers for developmental abnormalities in France. Only CREBBP variants associated with RSTS were included in this dataset, namely missense variants within the first 29 exons and protein-truncating variants. We included germline variations detectable from DNA extracted from whole blood. Along with 25 normal controls free of any neurodevelopmental condition, these samples whose status is perfectly and a priori known constitute the validation set and are described in Supplementary Table 1.
As a proof of concept, we also constituted a testing set, including 57 VUS carriers, as well as 8 “clinical hypothesis” cases, namely probands without definite molecular diagnostic despite a suggestive clinical presentation of a syndrome within one of the ten syndromes under investigation, as described in Supplementary Table 1.
All patients or legal representatives provided informed written consent for exome/genome analyses in a medical setting that contains a query on the use of residual samples for research. This genetic study was approved by our legal ethics committee.
DNA methylation analysis
Genomic DNA was extracted from whole blood and bisulfite converted. DNA methylation profile was then derived using Illumina’s Infinium EPIC array v1.0, in accordance with the manufacturer’s protocol. Carriers were randomly assigned a chip position while controls were homogeneously distributed over all rows of the chips. DNA methylation arrays were generated either by Diagenode SA, Liège, Belgium (n = 174) or by the Centre National de Recherche en Génomique Humaine (CNRGH), Evry, France (n = 22). Except for CNRGH samples which only contained AUTS18 cases and 3 CdL VUS carriers, unaffected controls and patients were evenly assigned over the beadchips and eight beadchip cells. These data were newly generated without overlap with original episignature training sets.
Raw IDAT data were processed and normalised using the Meffil R package [27]. This package efficiently handles large DNA methylation datasets. Briefly, probes which failed methylation detection (detection p value > 0.01) in more than 5% of samples were removed. Samples with >1% of failed probes or an outlier methylation distribution (methylation/unmethylation ≥ 3 s.d. from mean) were flagged. Three samples from the testing set (MRXSCJ_17, Sotos_12 and Sotos_18) failed these quality controls and were excluded from further steps. Remaining samples were functionally normalised together as advocated in the Meffil documentation, with adjustment on array, sentrix column and row, before computing β-values.
Several predictions derived from methylation values were added to the sample information table. Sex predictions were extracted from the standard meffil normalised object. No inconsistencies between reported and predicted sex were noted. Blood cell counts were estimated with the meffil.cell.count.estimates function. DNA methylation age was predicted with the DNAmAge function from the methyclock R package [28]. Among all available epigenetic clocks, the skinHorvath clock [29], which was trained on skin and blood 450 K samples, displayed the strongest correlation with actual age at inclusion on our dataset (Pearson correlation r = 0.91, 95% confidence interval [0.88–0.93]), and was thereby used as age predictor. For statistical analyses, we adjusted on age and sex predictions for consistency across samples with known or unknown age and sex.
Statistical analysis
We performed the following steps for each episignature, separately.
Literature data extraction
Episignature probes were not selected from our data. Instead, the probe list was retrieved from supplemental information of the corresponding publication (Table 1). All analyses regarding this episignature were then restricted to this specific list of probes. All probes are listed in Supplementary Table 3.
Case-control gap
To assess whether the episignature strength was reproduced on our dataset, we computed the proportion of CpG positions whose absolute average difference between cases and controls met the 5 and 10% thresholds that are typically required at discovery.
Adjustment for confounders
A linear regression model was fitted to adjust probe β-values on predicted age, sex and cell composition, which are all well-known confounders of methylation levels. Unless otherwise stated, residuals from this model were used in the following steps. Average residual methylation levels among pathogenic variant carriers and controls are given in Supplementary Table 3.
Evaluation of predictive abilities
Following a leave-one-out scheme, the kNN implementation from the “class” R package was used to predict the status (case or control) of every validation sample, case or control, using all remaining validation samples as training set. To guarantee high inter-syndrome specificity, a multiclass kNN was fitted to the full validation set simultaneously. The process was repeated for each validation sample. True positives and true negatives were then summarised into sensitivity and specificity estimations along with 95% confidence intervals based on an exact binomial distribution. To challenge the robustness of our results, we let the number k of nearest neighbours and the required level of consensus vary. Parameters ranged from “2/2”, perfect consensus between the two nearest neighbours, to “5/5”, perfect consensus between the five nearest neighbours. For 4 and 5 nearest neighbour predictions, because some syndromes display close signature profiles, we also allowed for the possibility of one discordant nearest neighbour (respectively “3/4” and “4/5”). KDM5C and ATRX genes being located on the X chromosome, it is expected that female carriers should not fully present the same episignature as male carriers. We therefore restricted sensitivity computations for these two genes to male samples. Inter-syndrome specificity was computed from other variant carriers.
Three subsidiary analyses were run to gain perspective:
-
Four original episignatures were accessible through the EpigenCentral (https://epigen.ccm.sickkids.ca/) open-access web-portal [25, 30]. We loaded our dataset onto the platform and followed user guide recommended practices.
-
To evaluate the gap between kNN and SVM predictors, an SVM algorithm (using the e1071 package default parameters) was fitted to residuals.
-
To evaluate the impact of age, sex and blood composition adjustment, a kNN was fitted on normalised betas without adjustment for age, sex and blood composition.
Visual representation
The reliability of case/control discrimination was visually inspected on the first two principal components as well as on a heatmap of DNA methylation residuals of episignature probes.
VUS classification
Finally, VUS and clinical hypothesis samples from the testing set were classified using the “3/4” kNN algorithm.
Results
Recruitment
A total of 101 samples from ten neuro-developmental disorders described in Table 1 along with 25 age and sex-matched controls free from neurodevelopmental disorders (validation set), 57 VUS carriers and 8 clinical hypothesis samples (testing set) passed meffil quality checks and were included in our evaluation.
Independent evaluation of episignature prediction accuracy
The reproducibility of episignatures was first assessed by computing the proportion of probes exceeding a 5 or 10% absolute average difference between cases and controls, as displayed in Table 1. Except ATRX, DNMT3A and both NSD1 signatures, all signatures displayed less than 50% positions above the 10% threshold. CdL, CHD8_2, CHD8_1 and CREBBP showed the lowest reproducibility, with <40% original positions reaching the 5% threshold, namely 39%, 35%, 22% and 14% respectively.
For all episignatures, the main available information is the set of probes used for prediction. Average β or Δβ profiles are often provided but they find themselves contaminated by batch effects. The prediction algorithm itself is never shared. In our efforts to validate most faithfully the performances of each episignature, we therefore combined signature probe information with our independent validation and testing datasets which could be handled robustly from raw datafiles. Namely, because methylation levels strongly depend on age, sex and cellular composition, all analyses and graphical representations were made from residual methylation levels, after adjustment for such major confounders. For every episignature, we combined a quantitative evaluation of prediction performances as obtained from kNN clustering with a visual inspection of DNA methylation profiles on principal components and heatmap. Sensitivity and specificity were estimated using a leave-one-out strategy on our independent dataset, guaranteeing the absence of overfitting and thereby unbiased estimates.
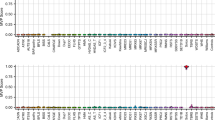
In all settings, our multiclass kNN prediction algorithm reached 100% specificity (95%CI [86–100%]), whatever the kNN parametrization, except for CHD8_2 which obtained a specificity of 96% [80–100%] under a 2/2 parametrization. As displayed on Fig. 1, five genes benefited from at least one signature reaching 100% sensitivity with multiple kNN configurations: ATRX, TBRS/DNMT3A, Kabuki/KMT2D, Sotos/NSD1, WDSTS/KMT2A_1 and 2. Robustness to kNN parametrization depended on the gene under investigation. A subset of episignatures showed good but highly variable sensitivities in the 40–90% range according to the kNN configuration used, namely CdL, CHARGE/CHD7, AUTS18/CHD8_1, and MRXCSJ/KDM5C_1 and 2. The sensitivity of these episignatures decreased as we increased the proportion of concordant nearest neighbours required for prediction. Two episignatures, AUTS18/CHD8_2 and RSTS/CREBBP, showed <40% sensitivities, whatever the configuration, with close to 0 sensitivity for highest numbers of concordant nearest neighbours. Overall, the “3/4” parametrization seemed to universally reach the best tradeoff given the sample size under study. Under such parametrization, inter-syndrome specificity was estimated at 100% [96%–100%] for all signatures except for CHD8_2 and both NSD1 signatures (99% [94%–100%]).

Panel (a) and (b) display sensitivities and specificities, respectively, according to various clustering parameters. Error bars indicate 95% confidence intervals based on a binomial distribution.
For most episignatures, sensitivity and specificity estimates from EpigenCentral, SVM predictor or non-adjusted methylation levels were similar to the results displayed in Fig. 1 (Supplementary Figs. 1–3). The main difference was observed for episignature CHD8_2, whose sensitivity rose to 100% [79.4%–100%] when using a SVM predictor or 93.8% [69.8%–99.8%] when working on non-adjusted methylation levels.
Visual inspection of PCA plots and heatmaps correlated well with sensitivity estimates. Three PCA plots and corresponding heatmaps, namely Kabuki/KMT2D_1, MXRSCJ/KDM5C_2 and RSTS/CREBBP, chosen to best illustrate the wide spectrum of episignature performances, are presented in Fig. 2. Graphical representations for other episignatures can be viewed in Supplementary Figs. 4–13. Notably, syndromes with 100% sensitivity displayed a separation of cases and controls by a large margin, while syndromes with intermediate and unstable sensitivity suffered from a strong heterogeneity among variant carriers, with a juxtaposition of what could be called extreme and milder DNA methylation profiles. The intermediate status of a subset of profiles is clearly visible on the heatmaps. AUTS18/CHD8_2 and RSTS/CREBBP episignatures showed incomplete separation between cases and controls.

Respectively for (A) Kabuki/KMT2D, (B) MXRSCJ/KDM5C and (C) RSTS/CREBBP, each panel displays the heatmap with simultaneous hierarchical clustering of validation and testing samples on the left. Blue indicates hypo-methylated positions while yellow indicates hyper methylated positions. First principal components are represented on the right. Percentage of explained variance is added in parenthesis on each axis.
The strength of episignatures was also reflected by the percentage of methylation variance within the signature that is explained by the first axis. Indeed, the separation between cases and controls was observed on the first and major axis of variation. On an episignature like Sotos/NSD1, this separation accounts for 86.7% of the variance. For MRXCSJ/KDM5C, this number falls down to 66.5%. On the RSTS/CREBBP episignature, the separation between cases and controls accounts for no more than 27.5% of the variance of methylation level residuals, leaving more than two thirds of the variance to unexplained factors or noise.
Surprisingly, two carriers of KDM5C class 4 and 5 variants appeared as outliers on both the heatmap and first principal components (MRXSCJ_7 and MRXSCJ_9, supplementary fig. 9). Sample identity was double checked by Sanger sequencing and a global status prediction of all validation samples in search for obvious sample swaps. The first outlier is MRXSCJ_7, carrying a de novo missense variant, NM_004187.3:c.593 G > A, p.(Arg198Gln), classified as likely pathogenic. The second outlier is MRXSCJ_9, carrying a small deletion NM_004187.3:c.645_657+5del, p.? (inheritance unknown), classified as pathogenic. ACMG-AMP classification of the variants was based on a collegial decision taking in silico pathogenicity predictions, mode of inheritance, clinvar co-occurences as well as phenotype concordance into account and is not questioned.
Among the ten syndromes under investigation, six had two distinct published episignatures. Except for AUTS18/CHD8, performances were somewhat similar between the two available signatures. In contrast, our data shows that the episignatures that we called AUTS18/CHD8_1 [18] clearly outperformed the other one (AUTS18/CHD8_2) [7].
Episignatures as a biomarker for VUS classification
Overall, we investigated 57 VUS carriers and 8 probands with a suggestive phenotype but negative exome sequencing. Sample characteristics and classification results are displayed in Supplementary Table 1. Several results deserve special attention.
Twelve VUS carriers showed methylation profiles compatible with the corresponding episignatures. From these, two samples had a discordant prediction between the two available episignatures. CHARGE_14 presented a positive episignature with the CHARGE/CHD7_1 [19] episignature but not by the CHARGE/CHD7_2 episignature [7]. The opposite was observed for the CHARGE_12 sample. Both segregated properly with cases in the PCA plot made from the CHARGE/CHD7_2 probes but separation between cases and controls was milder for this episignature, which can cause classification errors (Supplementary Fig. 7).
In fact, several episignatures, even among those with perfect separation and 100% sensitivity, found themselves challenged in practice by VUS with intermediate methylation profiles. Such a phenomenon was observed for sample Kabuki_11, which as a result presented a conflicted positive Kabuki/KMT2D_2 episignature but negative for Kabuki/KMT2D_1 (Fig. 2A). Although such profiles were expected and observed for female carriers of X-linked syndromes, two male KDM5C VUS carriers also presented intermediate profiles (Fig. 2B). Of course, this situation might arise from unobserved heterogeneity among cases, and such undecisive scenarios could be settled by the analysis of a larger dataset.
One proband carrying a pathogenic CHD7 complete duplication was included among VUS. This duplication presented a negative episignature, but PCA plot inspection revealed that it was projected on the opposite end of the first axis. This observation is consistent with the haploinsufficiency mediated pathogenicity in CHARGE syndrome [31].
Among patients with negative episignatures, Sotos_17 was later discovered to harbour a pathogenic variant in PTCH1, thus confirming its differential diagnosis.
Among clinical hypotheses, only one proband with a Sotos syndrome clinical phenotype harboured positive Sotos/NSD1 episignatures, suggesting that the patient actually presents this disorder and that further genetic analyses should be proposed to identify the causal variant.
Discussion
To our knowledge, we provide the first independent and unbiased evaluation of 16 episignatures spanning 10 neurodevelopmental disorders, in terms of predictive accuracy and robustness. All data were newly generated for this project, and none were included in previous training sets used for the selection of probes and detection of episignatures. For every signature, the multiclass and stringent kNN strategy guaranteed perfect specificity, with regard to both normal controls and other syndromes. This specificity estimation is based on 25 unaffected controls, but all matched to cases from a technical point of view. Resorting to large control sets from the GEO platform could have increased sample size but spurious differences between datasets would have biased our estimations. The perfect inter-syndrome specificity brings confidence to the interpretation of probands with a suggestive phenotype but with negative exome sequencing. On the other hand, sensitivity was highly heterogeneous among syndromes, with close results between kNN, SVM and EpigenCentral predictors. Combined with sensitivity estimates, visual inspection of PCA plots and heatmaps revealed that, essentially, episignatures could be split into three groups: (i) robust signatures ready for confident use in a diagnostic setting (ii) signatures of reasonable but unstable predictive abilities, facing challenges in practice (iii) weak signatures that are not ready for use in a diagnostic setting. The proportions of probes with large methylation gap between cases and controls seem highly evocative of this partition.
The first group includes ATRX, Sotos/NSD1, TBRS/DNMT3A and Kabuki/KMT2D robust signatures. Pathogenic variants in these four genes led to a perfect separation between cases and controls, displaying a robust 100% sensitivity. This observation is well documented in the literature, namely that Sotos and TBRS syndromes are known to cause a drastic hypo-methylated signature among carriers with large overlap between differentially methylated probe sets [7, 11, 20, 21, 26]. The similarities between signatures caused a slight risk of misclassification, even within our multiclass prediction framework. Increasing sample size should help decipher the biological meaning of these overlaps as well as reduce misclassifications. Inversely, the overlap between Kabuki/KMT2D_1 and Kabuki/KMT2D_2 is marginal (37 out of 153 and 221 probes respectively), reminding us that two distinct signatures can be equally powerful thanks to the highly correlated structure of CpG methylation levels. Besides, despite these perfect performances on the validation set, some VUS remained impossible to classify in practice within ATRX and KMT2D, raising the question of whether similar issues could arise in practice within NSD1 and DNMT3A genes, should we increase the number of VUS under investigation.
The second group consist of CdL, CHARGE/CHD7, AUTS18/CHD8_1, MRXCSJ/KDM5C, WDSTS/KMT2A, episignatures. These signatures showed reasonable sensitivity, making them effective biomarkers for VUS classification in theory, but intermediate profiles rendered sensitivity estimates dependent on classifier parametrization and interpretation complex in practice. Co-occurences of milder episignatures with milder phenotypes have been previously reported [16, 34,35,36]. Here, AUTS18_2 was the father of AUTS18_1, both carrying the same CHD8 pathogenic variant. While the daugther showed typical features of AUTS18/CHD8 (intellectual disability, autism spectrum disorder and macrocephaly), her father only displayed macrocephaly without any known neurodevelopmental features. Nevertheless, his methylation profile was without any doubt positive. That single observation does not support the hypothesis that milder phenotype inevitably coincides with intermediate profiles, at least regarding AUTS18/CHD8. We also identified two samples with opposite methylation patterns in KDM5C. Close attention revealed that the two variants affected the same exon. Works by Ugur et al. showed that amino acids 199 to 218 of KDM5C protein—in which these two variants are located—define a very specific functional domain, suggesting a potential domain-specific effect [32]. It is impossible to draw a firm conclusion on two occurences, but it is conceivable that pathogenic variants in this precise region should induce a distinct impact on DNA methylation. This scenario has already been described for ADNP which has a main episignature and a second one restricted to a specific protein domain [33]. More samples are required to confirm this hypothesis.
On the lower end of the spectrum are RSTS/CREBBP and AUTS18/CHD8_2 signatures. Our data advise against the use of these episignatures in a diagnostic setting. Reasons may be either biological or technical. Perhaps the methylation impact is not strong enough or cases suffer from a diversity which has not been understood yet. The low rate of positions reaching more than a 5% absolute methylation gap suggest these signatures suffer from winner’s curse and overfitting to a small discovery set (5 cases for AUTS18/CHD8_2). More recently, the same team reported in [11] an increased sample size of 28 CHD8 cases but no update was published regarding the probe list. In the same publication, eight new episignatures have been similarly trained on <5 samples. The SVM predictor improved the sensitivity of CHD8_2 by reinforcing the weight of the few CpG positions that remain differentially methylated on the validation dataset while discarding non-reproducible ones. This apparent increase in sensitivity is probably at the cost of some new overfitting.
A recent review on episignature provided several illustrations of the complexity of interpreting episignatures in practice, be it related to intermediate profiles with SMARCA2 or HNRNP examples or the existence of gene regions that evade the signature for EZH2 and SRCAP genes [34]. Our work suggest that these scenarios are much more common than could be expected. Without questioning the validity of corresponding episignatures, intermediate profiles and local exceptions demonstrate that episignatures cannot be used as automated binary tools. It is important that predictions should be challenged by careful visual and expert inspection. Precise interpretation of these complex episignatures requires further investigation of more samples to (i) confirm and understand the implications of diverse methylation profiles and (ii) investigate the existence of regions that may escape the signatures. A larger sample size would allow for genotype/methylation and phenotype/methylation correlation analyses to provide informed and accurate genetic counselling to carriers. As always in the molecular biology diagnostic process, biological and clinical expertise about the neurodevelopmental syndrome is mandatory to make an analysis reliable.
From a technical point of view, a limitation of our study is that all probes in the signature contributed equally to the prediction, contrary to other machine-learning models that combine probes with more flexibility. However, with only about ten samples per syndrome, kNN provided a more reasonable and robust approach which limits the introduction of a new level of overfitting. With larger sample sizes, other methodological options could be devised.
Code availability
Major code steps permitting to reproduce the analyses are given in Supplementary Information.
References
Wright CF, Fitzgerald TW, Jones WD, Clayton S, McRae JF, van Kogelenberg M, et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet. 2015;385:1305–14.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Kaplanis J, Samocha KE, Wiel L, Zhang Z, Arvai KJ, Eberhardt RY, et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature. 2020;586:757–62.
Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 6 févr. 2020;180:568–584.e23.
Aref-Eshghi E, Schenkel LC, Lin H, Skinner C, Ainsworth P, Paré G, et al. The defining DNA methylation signature of Kabuki syndrome enables functional assessment of genetic variants of unknown clinical significance. Epigenetics. 2017;12:923–33.
Aref-Eshghi E, Bend EG, Hood RL, Schenkel LC, Carere DA, Chakrabarti R, et al. BAFopathies’ DNA methylation epi-signatures demonstrate diagnostic utility and functional continuum of Coffin-Siris and Nicolaides-Baraitser syndromes. Nat Commun. 2018;9:4885.
Aref-Eshghi E, Kerkhof J, Pedro VP, DI Groupe F, Barat-Houari M, et al. Evaluation of DNA Methylation Episignatures for Diagnosis and Phenotype Correlations in 42 Mendelian Neurodevelopmental Disorders. Am J Hum Genet. 2020;106:356–70.
Cherik F, Reilly J, Kerkhof J, Levy M, McConkey H, Barat-Houari M, et al. DNA methylation episignature in Gabriele-de Vries syndrome. Genet Med. 2022;24:905–14.
Foroutan A, Haghshenas S, Bhai P, Levy MA, Kerkhof J, McConkey H, et al. Clinical Utility of a Unique Genome-Wide DNA Methylation Signature for KMT2A-Related Syndrome. Int J Mol Sci. 2022;23:1815.
Haghshenas S, Levy MA, Kerkhof J, Aref-Eshghi E, McConkey H, Balci T, et al. Detection of a DNA Methylation Signature for the Intellectual Developmental Disorder, X-Linked, Syndromic, Armfield Type. Int J Mol Sci. 2021;22:1111.
Levy MA, McConkey H, Kerkhof J, Barat-Houari M, Bargiacchi S, Biamino E, et al. Novel diagnostic DNA methylation episignatures expand and refine the epigenetic landscapes of Mendelian disorders. HGG Adv. 2022;3:100075.
Mirza-Schreiber N, Zech M, Wilson R, Brunet T, Wagner M, Jech R, et al. Blood DNA methylation provides an accurate biomarker of KMT2B-related dystonia and predicts onset. Brain. 2022;145:644–54.
Radio FC, Pang K, Ciolfi A, Levy MA, Hernández-García A, Pedace L, et al. SPEN haploinsufficiency causes a neurodevelopmental disorder overlapping proximal 1p36 deletion syndrome with an episignature of X chromosomes in females. Am J Hum Genet. 2021;108:502–16.
Rooney K, Levy MA, Haghshenas S, Kerkhof J, Rogaia D, Tedesco MG, et al. Identification of a DNA Methylation Episignature in the 22q11.2 Deletion Syndrome. Int J Mol Sci. 2021;22:8611.
Rouxel F, Relator R, Kerkhof J, McConkey H, Levy M, Dias P, et al. CDK13-related disorder: Report of a series of 18 previously unpublished individuals and description of an epigenetic signature. Genet Med. 2022;24:1096–107.
Schenkel LC, Kernohan KD, McBride A, Reina D, Hodge A, Ainsworth PJ, et al. Identification of epigenetic signature associated with alpha thalassemia/mental retardation X-linked syndrome. Epigenet Chrom. 2017;10:10.
Schenkel LC, Aref-Eshghi E, Rooney K, Kerkhof J, Levy MA, McConkey H, et al. DNA methylation epi-signature is associated with two molecularly and phenotypically distinct clinical subtypes of Phelan-McDermid syndrome. Clin Epigenet. 2021;13:2.
Siu MT, Butcher DT, Turinsky AL, Cytrynbaum C, Stavropoulos DJ, Walker S, et al. Functional DNA methylation signatures for autism spectrum disorder genomic risk loci: 16p11.2 deletions and CHD8 variants. Clin Epigenet. 2019;11:103.
Butcher DT, Cytrynbaum C, Turinsky AL, Siu MT, Inbar-Feigenberg M, Mendoza-Londono R, et al. CHARGE and Kabuki Syndromes: Gene-Specific DNA Methylation Signatures Identify Epigenetic Mechanisms Linking These Clinically Overlapping Conditions. Am J Hum Genet. 2017;100:773–88.
Levy MA, Relator R, McConkey H, Pranckeviciene E, Kerkhof J, Barat-Houari M, et al. Functional correlation of genome-wide DNA methylation profiles in genetic neurodevelopmental disorders. Hum Mutat. 2022;43:1609–28.
Choufani S, Cytrynbaum C, Chung BHY, Turinsky AL, Grafodatskaya D, Chen YA, et al. NSD1 mutations generate a genome-wide DNA methylation signature. Nat Commun. 2015;6:10207.
Kerkhof J, Squeo GM, McConkey H, Levy MA, Piemontese MR, Castori M, et al. DNA methylation episignature testing improves molecular diagnosis of Mendelian chromatinopathies. Genet Med. 2022;24:51–60.
Sadikovic B, Levy MA, Aref-Eshghi E. Functional annotation of genomic variation: DNA methylation episignatures in neurodevelopmental Mendelian disorders. Hum Mol Genet. 2020;29:R27–32.
Sadikovic B, Levy MA, Kerkhof J, Aref-Eshghi E, Schenkel L, Stuart A, et al. Clinical epigenomics: genome-wide DNA methylation analysis for the diagnosis of Mendelian disorders. Genet Med. 2021;23:1065–74.
Awamleh Z, Goodman S, Kallurkar P, Wu W, Lu K, Choufani S, et al. Generation of DNA Methylation Signatures and Classification of Variants in Rare Neurodevelopmental Disorders Using EpigenCentral. Curr Protoc. 2022;2:e597.
Chater-Diehl E, Goodman SJ, Cytrynbaum C, Turinsky AL, Choufani S, Weksberg R. Anatomy of DNA methylation signatures: Emerging insights and applications. Am J Hum Genet. 2021;108:1359–66.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–9.
Pelegí-Sisó D, de Prado P, Ronkainen J, Bustamante M, González JR. methylclock: a Bioconductor package to estimate DNA methylation age. Bioinformatics. 2021;37:1759–60.
Horvath S, Oshima J, Martin GM, Lu AT, Quach A, Cohen H, et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging. 2018;10:1758–75.
Turinsky AL, Choufani S, Lu K, Liu D, Mashouri P, Min D, et al. EpigenCentral: Portal for DNA methylation data analysis and classification in rare diseases. Hum Mutat. 2020;41:1722–33.
Lalani SR, Safiullah AM, Fernbach SD, Harutyunyan KG, Thaller C, Peterson LE, et al. Spectrum of CHD7 Mutations in 110 Individuals with CHARGE Syndrome and Genotype-Phenotype Correlation. Am J Hum Genet. 2006;78:303–14.
Ugur FS, Kelly MJS, Fujimori DG. Chromatin Sensing by the Auxiliary Domains of KDM5C Regulates Its Demethylase Activity and Is Disrupted by X-linked Intellectual Disability Mutations. J Mol Biol. 2023;435:167913.
Bend EG, Aref-Eshghi E, Everman DB, Rogers RC, Cathey SS, Prijoles EJ, et al. Gene domain-specific DNA methylation episignatures highlight distinct molecular entities of ADNP syndrome. Clin Epigenet. 2019;11:64.
Awamleh Z, Goodman S, Choufani S, Weksberg R. DNA methylation signatures for chromatinopathies: current challenges and future applications. Hum Genet. 2023. https://doi.org/10.1007/s00439-023-02544-2. Epub ahead of print.
Awamleh Z, Choufani S, Cytrynbaum C, Alkuraya FS, Scherer S, Fernandes S, et al. ANKRD11 pathogenic variants and 16q24.3 microdeletions share an altered DNA methylation signature in patients with KBG syndrome. Hum Mol Genet. 2023;32:1429–38.
Oexle K, Zech M, Stühn LG, Siegert S, Brunet T, Schmidt WM, et al. Episignature analysis of moderate effects and mosaics. Eur J Hum Genet. 2023;31:1032–9.
Acknowledgements
This work was performed in the framework of FHU-G4 Génomique.
Funding
DNA methylation chips were generated thanks to Fondation de l’Avenir, Fondation Deniker and Cerveau Progrès fundings. The CEA/CNRGH sequencing platform was supported by the France Génomique National infrastructure, funded as part of the « Investissements d’Avenir » program managed by the Agence Nationale pour la Recherche (contract ANR-10-INBS-09).
Author information
Authors and Affiliations
Contributions
TH, GN, OG, DC and CC designed this work. All authors played a role in acquiring data, either by recruiting samples, generating methylation data or running statistical analyses. TH, FL, GN, CC played an important role in interpreting the results. TH, CC drafted the paper, all authors contributed to the revision of the paper and approved the final version. All authors agreed to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study was approved by the CERDE, which is the local ethics committee.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Husson, T., Lecoquierre, F., Nicolas, G. et al. Episignatures in practice: independent evaluation of published episignatures for the molecular diagnostics of ten neurodevelopmental disorders. Eur J Hum Genet 32, 190–199 (2024). https://doi.org/10.1038/s41431-023-01474-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-023-01474-x
