
Abstract
Orofaciodigital syndrome is a distinctive subtype of skeletal ciliopathies. Disease-causing variants in the genes encoding the CPLANE complex result in a wide variety of skeletal dysplasia with disturbed ciliary functions. The phenotypic spectrum includes orofaciodigital syndrome and short rib polydactyly syndrome. FUZ, as a part of the CPLANE complex, is involved in intraflagellar vesicular trafficking within primary cilia. Previously, the variants, c.98_111+9del and c.851G>T in FUZ were identified in two individuals with a skeletal ciliopathy, manifesting digital anomalies (polydactyly, syndactyly), orofacial cleft, short ribs and cardiac defects. Here, we present two novel variants, c.601G>A and c.625_636del in biallelic state, in two additional subjects exhibiting phenotypic overlap with the previously reported cases. Our findings underscore the association between biallelic loss of function variants in FUZ and skeletal ciliopathy akin to orofaciodigital syndrome.
Similar content being viewed by others


Introduction
Skeletal ciliopathies are a diverse group of rare disorders [1]. These conditions are caused by either a defect in the formation of cilia or a dysfunction of centrosome or cilia. They are recognized by a broad range of phenotypes: short ribs, short limbs, short digits, polydactyly, trident ilia, cone shaped epiphyses and cupped metaphases. They also exhibit pleiotropy and affect the craniofacial, nervous, cardiac, gastrointestinal, and genitourinary systems [2,3,4,5].
The primary cilia is a non-motile protrusion from the cell surface of most mammalian cells. It is involved in cell signaling and development. The assembly of cilia requires synchronized involvement of several regulatory complexes and signaling proteins [6]. One of the essential yet least understood complexes is CPLANE (ciliogenesis and planar polarity effector). The core CPLANE complex consists of INTU (Inturned Planar Cell Polarity Protein), FUZ (Fuzzy Planar Cell Polarity Protein) and WDPCP (WD Repeat Containing Planar Cell Polarity Effector), which also strongly interacts with CPLANE1 and RSG1 (CPLANE2) [7]. Perturbation of the expression of these genes might cause disruption of ciliogenesis and result in skeletal ciliopathies (Short rib‐polydactyly syndrome (SRPS), INTU‐related (MIM# 617925), Congenital heart defects, hamartomas of tongue, and polysyndactyly, WDPCP-related (MIM# 217085), Joubert syndrome 17, CPLANE1-related (MIM# 614615) and Orofaciodigital syndrome VI, CPLANE1-related (MIM# 277170). The phenotypic spectrum is wide and includes short rib thoracic dysplasia and orofaciodigital syndrome.
Recently, FUZ has been implicated in vesicular trafficking within primary cilia [7, 8]. FUZ plays a critical role in transporting protein necessary for cilium assembly and cell signaling such as Wnt, FGF, hedgehog signaling pathways. The intricate network of multiple signaling pathways and interactions among germ layers and neural crest cells is crucial during the process of organogenesis and other embryonic developmental processes. Dysregulation of these networks due to disruption in ciliary function can contribute to various diseases. It has been observed that defects in FUZ are associated with severe neural tube defects, craniosynostosis, cardiac abnormalities, or skeletal dysplasia [8,9,10,11,12]. Only two patients have been described until now where biallelic loss of function variants in FUZ were expected to result in a skeletal ciliopathy [4]. Herein, we describe two Indian patients carrying biallelic variants in FUZ with digital anomalies, orofacial cleft, short ribs and cardiac defects resembling orofaciodigital syndrome.
Methods
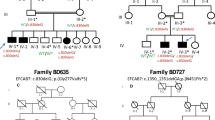
Two families were ascertained for clinical testing (Figs. 1 and 2) in view of skeletal dysplasia. Genomic DNA was isolated from the blood using QIAmp DNA Blood Mini Kit (Cat# 51106, QIAGEN, USA). Exome sequencing was performed using the protocol described previously [13]. Furthermore, data was annotated by ‘Annotate Variation (ANNOVAR)’ [14] and our in-house scripts [15]. The filtered variants were then analyzed using relevant genetic databases and coding/noncoding variant pathogenicity predictors. Sanger sequencing was carried out for the validation and segregation of the rare disease-causing variants identified in the families (Fig. 1). Homozygous regions for a minimum size of 1 Mb from the exome sequencing data of the probands were identified using AutoMap [16]. The variants were described according to HGVS nomenclature, using reference sequences NM_025129.5 (GRCh38) and NP_079405.2. Both the variants are submitted to ClinVar (SUB13866645 and SUB13866677).

Sanger sequencing chromatograms show segregation of these variants in both families.

Short and broad thumbs and clinodactyly of 5th finger were noted (A and C). Preaxial and postaxial polysyndactyly were observed in the right foot (B and E). Chest radiograph is unremarkable (D).
Results
Family 1
The proband 1 is a two-years-nine-months old girl. Her birth weight was 2.8 kg (−1.19 SD). A congenital heart defect was noted during the antenatal scan. Her development and speech were normal. Her height was 94 cm (+0.37 SD), weight 10.5 kg (−2.24 SD) and head circumference 46 cm (−1.51 SD) at the time of examination. She had a prominent forehead, medial flaring of the eyebrow, low set ears with prominent antihelix, broad nasal bridge and narrow chest. Short and broad thumbs and clinodactyly of 5th fingers were noted. Additionally, bilateral preaxial polysyndactyly was evident in feet and postaxial polysyndactyly in the right foot. She had a partial atrioventricular canal defect (AVCD) that was surgically corrected. The clinical and radiological findings of proband 1 are described in Fig. 2.
Her elder sister died at the age of 1.5 years and was found to be similarly affected. A cardiac evaluation had revealed a complete AVCD in her. Parents informed that she had bilateral cleft lip and palate, clinodactyly of the right 5th fingers and medially deviated, broad great toes. Her DNA was not available for testing.
Exome sequencing revealed a novel homozygous missense variant (c.601G>A, p.(Glu201Lys) in exon 6 of FUZ. Sanger sequencing confirmed the presence of this variant in heterozygous state in her parents (Fig. 1). This variant is observed in four individuals in the gnomAD database (v2.1.1) in heterozygous state with an allele frequency of 0.0000131. MutationTaster, SIFT, and Polyphen predicted this variant to be disease causing and it has a CADD phred score of 25, suggesting it to be deleterious. The multiple sequence alignment tool predicted wild type amino acid to be conserved in several species (Supplementary Fig. 1).
Family 2
The parents of proband 2 were clinically unaffected and had: a five-year-old healthy son, a second trimester miscarriage at 16 weeks (no diagnosis), third pregnancy terminated at 19 weeks due to atrioventricular septal defect (AVSD), single umbilical artery and presence of polydactyly. In the fourth conception, at 25 years of the mother’s age, the antenatal scan showed a right cleft lip, polydactyly (seven digits in both hands and feet), bilateral duplicated hallux and AVSD in proband 2 at 19 weeks of gestation. The pregnancy was medically terminated, and the genomic DNA was tested.
Compound heterozygous variants were identified in exon 6 of FUZ: a in-frame deletion variant inherited from the mother c.625_636del, p.(Val209_Leu212del) and a missense variant c.601G>A, p.(Glu201Lys) inherited from the father (Fig. 1). Segregation analysis could not be performed in the healthy sibling as his DNA sample was unavailable for testing. The variant c.601G>A was also found in the unrelated family 1. The variant c.625_636del was absent in gnomAD database (v2.1.1) and results in in-frame deletion leading to change in protein length. The clinical and molecular findings of both probands and those reported earlier are summarized in Table 1. The variant c.601G>A resides in an autozygous region region in family 1 and shares the same haplotype with family 2 indicating a shared ancestry (Supplementary Tables 1 and 2).
Discussion
In this study, we describe two Indian families with phenotypes suggestive of a skeletal ciliopathy implicated by biallelic variants in FUZ. Both the families had multiple affected fetuses individuals, though genetic testing was performed only in the probands and parents.
FUZ has recently been associated with skeletal ciliopathy in humans [4, 8]. Digital anomalies (polydactyly, syndactyly), orofacial cleft, short ribs and cardiac defects are the major clinical findings in the patients (including this report) which has resemblance to the genetically heterogeneous orofaciodigital syndromes (Table 1). Four variants (two missense and two predicted loss of function), in biallelic state in FUZ in these patients suggest loss of function of FUZ as the likely cause of the skeletal phenotype in these families (Table 1 and Supplementary Fig. 2).
Possible role for FUZ in skeletal involvement was discovered earlier when Fuz knockout mice showed severe developmental abnormalities comprising craniofacial defects, malformed sternum, ribs and long bones, polydactyly, and incompletely penetrant rostral neural tube closure defects [10, 11, 17]. Currently known functions of CPLANE complex and the phenotypes that result from deficiency of the component proteins (listed above in the introduction) predict a skeletal ciliopathy for the deficiency of FUZ too, consistent with our observations.
Zhang et al., described a bi-allelic splice variant c.98_111+9del leading to frameshift deletion and likely loss of function of FUZ. This variant led to the prenatal death of the patient. The phenotypes of the patient comprised a long, narrow chest, moderately short ribs, short long bones, and extreme polydactyly of all four limbs, ventricular septal defect, hypoplastic kidneys, midline facial cleft, thickened nuchal fold and dilated third ventricle [4].
Similarly, in-frame deletion variant c.625_636del, detected in compound heterozygous state in family 2 is predicted to cause in-frame deletion. The impact of the variant, whether it leads to a less functional protein with an internal alteration or results in instability and loss of the gene product, remains unknown. The predicted consequence of in-frame deletion is retention of an altered protein product.
The missense variant c.851G>T, p.(Arg284Leu) was also reported by Zhang et al. in 2018 in a patient who had features of asphyxiating thoracic dystrophy/short-rib thoracic dysplasia with polydactyly, though they did not identify a second variant [4]. Another biallelic variant, p.(Arg284pro) with a different amino acid change at the same position was noted by Barrel et al. in 2021 in a pair of twins who had craniosynostosis and dilatation of the lateral brain ventricles with no other skeletal manifestations [8]. Functional assays demonstrated that this variant could partially rescue Fuz mutant ciliogenesis but showed impaired hedgehog signaling transduction. They further established that Fuz mutant osteoblasts had increased potential of mineralization compared to the wildtype which might affect craniofacial ossification. This variant was located in domain 2 of FUZ, which is upstream of C-terminal domain. C-terminal domain is predominantly involved in vesicular trafficking, critical for ciliogenesis [17, 18]. Association of heterozygous variants in FUZ with neural tube defect observed by Seo et al. has not been substantiated yet by other studies [12].
Both the missense p.(Glu201Lys) and in-frame deletion p.(Val209_Leu212del), may have a less deleterious effect than a null variant c.98_111+9del. which might explain the milder skeletal phenotype of orofaciodigital syndrome observed in surviving probands in the study. This is along the expected lines in the spectrum of skeletal ciliopathies where boundaries between the several phenotypic descriptions are indistinct. For instance, biallelic variants in CEP120 results in Joubert syndrome 31 and severe short-rib thoracic dysplasia type 13 while spectrum of KIAA0753 related conditions extends to Joubert syndrome 38, orofaciodigital syndrome XV andshort-rib thoracic dysplasia 21without polydactyly [1, 19, 20]. Further clinical and mechanistic studies would help in understanding the genetic defects and their relationship to the phenotype. In conclusion, this study provides clinical and genetic evidence for autosomal recessive FUZ-related skeletal ciliopathy.
Web resources
PRIMER 3v.4.1.0, http://primer3.ut.ee/
Ensembl, https://asia.ensembl.org/index.html
NCBI, https://www.ncbi.nlm.nih.gov/
Mutation Taster, http://www.mutationtaster.org/
OMIM, https://www.omim.org/
gnomAD, https://gnomad.broadinstitute.org/
ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/
HGMD, http://www.hgmd.cf.ac.uk/ac/search.php
Mutalyzer, https://mutalyzer.nl/
Data availability
Additional data supporting the study are available from the corresponding author upon reasonable request.
References
Unger S, Ferreira CR, Mortier GR, Ali H, Bertola DR, Calder A, et al. Nosology of genetic skeletal disorders: 2023 revision. Am J Med Genet A. 2023;191:1164–209.
Martín-Salazar JE, Valverde D. CPLANE Complex and Ciliopathies. Biomolecules. 2022;12:847.
Handa A, Voss U, Hammarsjö A, Grigelioniene G, Nishimura G. Skeletal ciliopathies: a pattern recognition approach. Jpn J Radio. 2020;38:193–206.
Zhang W, Taylor SP, Ennis HA, Forlenza KN, Duran I, Li B, et al. Expanding the genetic architecture and phenotypic spectrum in the skeletal ciliopathies. Hum Mutat. 2018;39:152–66.
Lai B, Jiang H, Gao Y, Zhou X. Skeletal ciliopathy: pathogenesis and related signaling pathways. Mol Cell Biochem. 2023. Available from: https://doi.org/10.1007/s11010-023-04765-5.
Sánchez I, Dynlacht BD. Cilium assembly and disassembly. Nat Cell Biol. 2016;18:711–7.
Toriyama M, Lee C, Taylor SP, Duran I, Cohn DH, Bruel AL, et al. The ciliopathy-associated CPLANE proteins direct basal body recruitment of intraflagellar transport machinery. Nat Genet. 2016;48:648–56.
Barrell WB, Adel Al-Lami H, Goos JAC, Swagemakers SMA, van Dooren M, Torban E, et al. Identification of a novel variant of the ciliopathic gene FUZZY associated with craniosynostosis. Eur J Hum Genet. 2022;30:282–90.
Arrigo AB, Lin JHI. Endocytic Protein Defects in the Neural Crest Cell Lineage and Its Pathway Are Associated with Congenital Heart Defects. Int J Mol Sci. 2021;22:8816.
Tabler JM, Barrell WB, Szabo-Rogers HL, Healy C, Yeung Y, Perdiguero EG, et al. Fuz Mutant Mice Reveal Shared Mechanisms between Ciliopathies and FGF-Related Syndromes. Dev Cell. 2013;25:623–35.
Zhang Z, Wlodarczyk BJ, Niederreither K, Venugopalan S, Florez S, Finnell RH, et al. Fuz regulates craniofacial development through tissue specific responses to signaling factors. PLoS One. 2011;6:e24608.
Seo JH, Zilber Y, Babayeva S, Liu J, Kyriakopoulos P, De Marco P, et al. Mutations in the planar cell polarity gene, Fuzzy, are associated with neural tube defects in humans. Hum Mol Genet. 2011;20:4324–33.
Girisha KM, von Elsner L, Neethukrishna K, Muranjan M, Shukla A, Bhavani GS, et al. The homozygous variant c.797G>A/p.(Cys266Tyr) in PISD is associated with a Spondyloepimetaphyseal dysplasia with large epiphyses and disturbed mitochondrial function. Hum Mutat. 2019;40:299–309.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Kausthubham N, Shukla A, Gupta N, Bhavani GS, Kulshrestha S, Das Bhowmik A, et al. A dataset of variants derived from 1455 clinical and research exomes is efficient in variant prioritization for early-onset monogenic disorders in Indians. Hum Mutat. 2021;42:e15–61.
Quinodoz M, Peter VG, Bedoni N, Royer Bertrand B, Cisarova K, Salmaninejad A, et al. AutoMap is a high performance homozygosity mapping tool using next-generation sequencing data. Nat Commun. 2021;12:518.
Gray RS, Abitua PB, Wlodarczyk BJ, Szabo-Rogers HL, Blanchard O, Lee I, et al. The planar cell polarity effector Fuz is essential for targeted membrane trafficking, ciliogenesis and mouse embryonic development. Nat Cell Biol. 2009;11:1225–32.
Brooks ER, Wallingford JB. Control of vertebrate intraflagellar transport by the planar cell polarity effector Fuz. J Cell Biol. 2012;198:37–45.
Hammarsjö A, Wang Z, Vaz R, Taylan F, Sedghi M, Girisha KM, et al. Novel KIAA0753 mutations extend the phenotype of skeletal ciliopathies. Sci Rep. 2017;7:15585.
Shaheen R, Schmidts M, Faqeih E, Hashem A, Lausch E, Holder I, et al. A founder CEP120 mutation in Jeune asphyxiating thoracic dystrophy expands the role of centriolar proteins in skeletal ciliopathies. Hum Mol Genet. 2015;24:1410–9.
Acknowledgements
We thank the patients and their families for participating in the study.
Funding
This work was partially supported by DBT/Wellcome Trust India Alliance Early Career Clinical and Public Health fellowship (IA/CPHE/20/1/505226) to Dhanya Lakshmi Narayana and Joint CSIR-UGC NET Junior Research Fellowship awarded by Human Resource Development Group under Council of Scientific and Industrial Research (CSIR), Government of India, to Swati Singh (08/028(0002)/2019-EMR-I). Open access funding provided by Manipal Academy of Higher Education, Manipal.
Author information
Authors and Affiliations
Contributions
SSingh: data curation and writing the first draft. SN and DLN: concept and clinical evaluation of the patients. CC: molecular diagnosis. SSalvankar: exome data analysis. KMG: concept, clinical evaluation of the patients, and writing the first draft. All authors have edited the manuscript drafts and revisions and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests. KMG holds shares and is a director of Suma Genomics Private Limited that has interests in clinical diagnostics.
Ethics approval
Consents for genetic testing on clinical basis were obtained from both families. Informed research consent was obtained for the second family, as approved by the Institutional Ethics Committee, Kasturba Medical College and Hospital, Manipal (IEC: 363/2020).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Singh, S., Nampoothiri, S., Narayanan, D.L. et al. Biallelic loss of function variants in FUZ result in an orofaciodigital syndrome. Eur J Hum Genet (2024). https://doi.org/10.1038/s41431-024-01619-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41431-024-01619-6
