
Abstract
EPM1 is the most common form of Progressive Myoclonus Epilepsy characterized by late-childhood onset, ever-worsening and disabling myoclonus, seizures, ataxia, psychiatric disease, and shortened lifespan. EPM1 is caused by expansions of a dodecamer repeat sequence in the promoter of CSTB (cystatin B), which dramatically reduces, but does not eliminate, gene expression. The relatively late onset and consistent presence of a minimal amount of protein product makes EPM1 a favorable target for gene replacement therapy. If treated early, these children’s normally developed brains could be rescued from the neurodegeneration that otherwise follows, and their cross-reactive immunological material (CRIM) positive status greatly reduces transgene related toxicity. We performed a proof-of-concept CSTB gene replacement study in Cstb knockout mice by introducing full-length human CSTB driven by the CBh promoter packaged in AAV9 and administered at postnatal days 21 and 60. Mice were sacrificed at 2 or 9 months of age, respectively. We observed significant improvements in expression levels of neuroinflammatory pathway genes and cerebellar granule cell layer apoptosis, as well as amelioration of motor impairment. The data suggest that gene replacement is a promising therapeutic modality for EPM1 and could spare affected children and families the ravages of this otherwise severe neurodegenerative disease.
Similar content being viewed by others


Introduction
Progressive Myoclonus Epilepsy Type 1 (EPM1), historically known as Unverricht-Lundborg disease, is an ultra-rare disease, but the most common form of inherited progressive myoclonus epilepsy (PME), which is characterized by myoclonus, epilepsy, and progressive neurologic deterioration [1]. Patients become symptomatic between the ages of 6 and 16 years with stimulus-sensitive, action-activated myoclonus, and tonic-clonic seizures. Over time, patients develop ataxia, lack of coordination, intention tremor, and dysarthria. EPM1 is caused by mutations in the CSTB gene and is inherited in autosomal recessive manner. CSTB encodes cystatin B, a cysteine protease inhibitor. Despite some progress in understanding the biological functions of cystatin B, a unifying disease mechanism remains unknown. By far the most common mutation in EPM1 is a dodecamer repeat expansion in the CSTB gene (upstream to the promoter), which leads to a reduction in CSTB expression [2]. EPM1 patients retain only ~10% of Cystatin B enzymatic activity [3]. In contrast, patients completely lacking CSTB protein present with an altogether different phenotype, namely a neonatal developmental encephalopathy with a severe disease course [4, 5]. This indicates a spectrum of disease whereby severity correlates with CSTB protein expression (with higher amounts of protein leading to lesser severity), suggesting that even small amounts of CSTB expression should lead to an improvement in disease symptoms. There are no disease-modifying therapies currently available for EPM1.
EPM1 is an attractive gene replacement therapy candidate for several reasons. First, there is a significant lag between early treatable symptoms and intractability, allowing time for intervention and a window to retain quality of life. Second, the disease is caused preponderantly by a single mutation leading to inadequate protein amount rather than a complete absence of the protein. This is important, as the gene defect should not elicit an immune response to an unfamiliar protein. Third, recent results suggest that CSTB is a secreted protein, and thus expression in the subset of brain cells transduced by the viral vector may be sufficient to rescuing adjacent cells and ultimately the entire brain [6]. Lastly, there is a mouse model that presents with many of the pathological features seen in EPM1 available for pre-clinical testing [7].
Adeno-Associated Virus (AAV) is a non-pathogenic viral vector that can deliver a transgene of choice to many different tissues and has become the most prominent vector used for gene therapy [8, 9]. In this report, we perform a proof-of-concept gene replacement therapy study for EPM1. We package human CSTB driven by the CBh promoter into AAV serotype 9 (AAV9), which we deliver via intrathecal lumbar puncture to the Cstb−/− mouse model. We show pathological improvements in the brain and functional improvements in the mice when treated at early or late stages during disease progression.
Results
AAV-CSTB brain transduction and protein expression
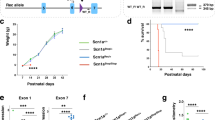
The absence of CSTB protein leads to a severe neurodegenerative disease that affects the entire brain but leaves the remainder of the body relatively unharmed in the mouse model [7, 10, 11]. To attempt to alleviate brain pathology, three cohorts of Cstb−/− mice were injected intrathecally with either AAV-CSTB (human CSTB) or PBS vehicle control. Both cohorts 1 and 2 were treated at postnatal day 21 (P21; early time point); cohort 1 was sacrificed at 2 months and cohort 2 at 9 months of age. Cohort 3 was treated at P60 (late time point) and sacrificed at 9 months (Fig. 1).

a AAV9 vector with a CBh promoter driving expression of human CSTB (AAV-CSTB). B AAV-CSTB or PBS administrated to three cohorts of Cstb−/− mice intrathecally. Cohort 1 and 2 were both injected at P21 (early time point) and brains examined at 2 and 9 months, respectively. Mice in cohort 3 were injected at p60 and brains examined at 9 months. Wild type (WT) littermates were left untreated in each cohort and used as controls.
To confirm transduction across the brain and assess distribution, we performed western blotting and immunohistochemistry with anti-CSTB antibody that recognizes human, but not mouse, CSTB [12]. In all cohorts, human CSTB protein was only present in the AAV-CSTB treated group where it showed broad distribution across the brain, with greater prominence in the cerebellum, medulla, pons, and midbrain. Interestingly, the expression level was more robust in later-sacrificed cohorts 2 and 3 compared to cohort 1 (Fig. 2).

AAV9-CSTB and PBS-treated representative mouse brain sections (lower right insets) from a cohort 1, c cohort 2, and e cohort 3, were subjected to immunohistochemistry using a human specific CSTB antibody. Scale bar is 1 mm. Western blot analysis was performed using brain tissue from AAV9-CSTB and PBS-treated mice of cohorts 1(b), 2 (d), and 3 (f).
AAV-CSTB gene replacement reduces early-onset neuroinflammation
One of the hallmarks of EPM1 pathogenesis in the mouse model is early onset neuroinflammation. Several immune markers including Cxcl1, Cxcl10, Cxcl13, Gfap, and Iba1 are upregulated in the Cstb−/− brain. Immune marker upregulation is especially pronounced at early stages of the disease where it follows an early microglial activation pattern, precedes neuronal loss, and diminishes through the late stages of the disease [13,14,15,16]. We quantified the expression of the above markers in wild type (WT), AAV-CSTB or PBS treated mice to assess the effect of gene therapy. In cohort 1, expression of three of these genes, Cxcl13, Iba1 and Gfap, was significantly reduced after AAV-CSTB treatment at 2 months of age. Levels of Cxcl1 and Cxcl10 also decreased though not to levels that reached statistical significance (Fig. 3a).

Relative mRNA expression levels of neuroinflammation markers Cxcl1, Cxcl10, Cxcl13, Iba1, and Gfap, were analyzed in cohorts 1 (a), 2 (b), and 3 (c) by qRT-PCR. Cohort 1, N = 15, per group. Cohort 2 and 3, WT (N = 13), PBS (N = 20), AAV-CSTB (N = 15). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. ns denotes non-significant.
At 7–9 months of age, the Cstb−/− mouse brain exhibited much lower levels of immune marker upregulation compared to 2 months of age. Treatment with AAV-CSTB appeared to reduce immune marker upregulation in cohorts 2 and 3, however, the reduction was not statistically significant except for Gfap in cohort 3 (Fig. 3b, c).
AAV-CSTB gene replacement decreases cerebellar granule cell death
Cstb−/− mouse brains undergo neurodegeneration, a large portion of which occurs even as the brain is developing, i.e., in the first three months of life [17, 18]. To determine whether AAV-CSTB rescues the loss of brain tissue, we measured brain mass at sacrifice in the three cohorts and found no differences between AAV-CSTB and PBS treated mice (Fig. 4a, b).

The weight of ground brain tissue from one hemisphere of a. cohort 1 (p21) at the age of 2 months and b. cohort 2 (p21) and 3 (p60) at the age of 7–9 months, was measured. Data represent the means of WT (N = 14 for cohort 1 and N = 12 for cohort 2 & 3), PBS (N = 13 for cohort 1 and N = 20 for cohort 2 and 3), AAV-CSTB p21 (N = 13 for cohort 1 and N = 15 for cohort 2 and 3) ± SEM. c Apoptotic bodies were detected in cerebellar sections using TUNEL assays. Propidium iodide (PI) was used for counterstaining. d Quantitative analysis for the number of apoptotic bodies in cerebellar sections is shown. Scale bars are 1 mm. Data represent the mean of WT (N = 16), PBS (N = 11) and AAV-CSTB (N = 15) ± SEM. ****p < 0.0001. ns denotes non-significant.
The neurodegeneration in Cstb−/− mice is apoptotic, which is best characterized in the cerebellum with the presence of abundant quantifiable apoptotic cells in the granule cell layer during the first three months of life. These apoptotic bodies decrease dramatically in subsequent months [7, 10]. We performed a TUNEL assay on paraffin-embedded cerebellar sections to compare cerebellar granule cell apoptosis. AAV-CSTB treated mice showed greater than 50% fewer apoptotic bodies compared to PBS-treated mice (Fig. 4c, d).
AAV-CSTB gene replacement improves ataxia
Starting from 6 months of age, Cstb−/− mice show mild signs of ataxia when challenged to walk on uneven surfaces, and lose their ability to remain on both the still and rotating rotarods as they age [1, 7]. These deficits are most prominent on the rotating rods (Fig. 5).

Cohort 2 (p21) and cohort 3 (p60) were subjected to both stationary and rotating (2 rpm) rotarods. Data represent the means of WT (N = 18), PBS (N = 21), and AAV-CSTB (N = 14) ± SEM for cohort 1, and WT (N = 18), PBS (N = 21) and AAV-CSTB (N = 15) ± SEM for cohort 2. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. ns denotes non-significant.
To assess the effects of gene replacement therapy on the above behavioral phenotype, 7–9 months old mice (from cohorts 2 and 3) were tested for ataxia using a rotating-rod apparatus. As mentioned, latency to fall from the stationary rod was significantly, though modestly, different between PBS and WT groups in cohorts 2 and 3. AAV-CSTB group showed a trend toward improvement, which did not reach statistical significance (Fig. 5). On the other hand, latency to fall from the rotating rod was greatly reduced in the PBS group and significantly increased with treatment (Fig. 5). These effects were the same in mice treated at postnatal days 21 or 60.
Discussion
EPM1 is an epilepsy with subtle onset that evolves into explosive intractable continuous myoclonus during the second and third decades of life. This is accompanied by convulsive seizures and rapidly worsening ataxia leading to wheelchair dependence. Symptoms somewhat diminish in intensity after the third decade but persist throughout the length of a reduced lifespan [19]. Given the severity, along with the neurodegenerative nature of the disease, the need for a treatment is acute. Lack of a treatment is also a high concern because relative to similarly severe conditions, EPM1 prevalence is high in certain countries (e.g. reaching 2/100,000 in Finland) [19, 20]. This is not merely due to a genetic founder effect. The causative mutation on at least one allele in all patients with EPM1 is a dodecamer repeat expansion that arises from a human-specific two or three-copy dodecamer repeat in the CSTB gene promoter [3]. The particularity of the location of this repeat sequence, and its predisposition to expand by virtue of its repetitive nature [21], suggest that EPM1 will continue to recur at this relatively high incidence. As a comparison, a differential diagnosis, the Lafora PME, only has an incidence of ~2/1,000,000, even in societies with high rates of consanguinity and despite the fact that it can be caused by recessively inherited mutations in two independent and much larger genes [22].
Numerous functions have been attributed to cystatin B, the first, where it derived its name, is as an inhibitor of lysosomal cysteine proteases [23]. Other functions include regulation of microglial activation and (neuro) inflammation, organizing synaptic transmission, interneuron migration and chromatin structure during neural stem cell renewal and differentiation, prevention of oxidative stress-induced cell death, and alteration of malignant characteristics of cancers [15, 24,25,26,27,28,29]. Consistent with its functional plurality, the CSTB protein has been documented in multiple subcellular locations, including at the lysosomal surface, in the lysosome, nucleus, and cytosol, and most recently, secreted extracellularly [6, 24, 27, 30]. The specific function of CSTB that contributes most prominently to EPM1 is unknown, which has been an impediment to developing disease-modifying therapies. A gene-based approach is therefore favored. Recent evidence that the product can be secreted [26], and thus is potentially able to impact cells beyond those transduced by the gene therapy vector, offers further support for pursuing this modality.
Another favorable circumstance is the intactness of affected children prior to disease onset. Early clinical or pre-symptomatic gene replacement could potentially protect the brain while it is still minimally affected, and neurons remain spared. Finally, the fact that all patients have a small amount of CSTB still produced diminishes concerns of immune intolerance to the delivered gene product.
The Cstb−/− mice are not a genetic model of EPM1. They are a genetic model of a disease caused by a complete absence of CSTB that is characterized by very severe early infantile encephalopathy and death in the rare cases where such children are even born [4, 5]. As such, phenotypic corrections in this mouse model point to a likely even greater impact in the less severe hypomorphic EPM1 phenotype. In the present work, to replicate the EPM1 clinical situation (late-childhood onset) we treated the mice at the relatively advanced ages of p21 and p60. At these ages, CSF-interstitial space barriers are mature and viral distribution is more restricted compared to gene therapies delivered in the neonatal period [31]. As such, our experimental design had a two-fold high bar, namely treating both later during disease progression and in a model of a much earlier-onset and much more severe disease than the target condition.
Despite this high bar, we were able to show that CSTB gene replacement reduces neuronal apoptosis, neuroinflammation and ataxia. Brain weight was not rescued. One explanation for the lack of brain weight rescue could be that a large portion of brain weight loss (likely reflecting neurodegeneration) had already occurred in these mice by 2 months of age (Fig. 4a, b; see also references) [10, 18]. Our earliest gene therapy treatment in mice was administered at p21, and maximal and stable protein production requires 3–4 weeks [32,33,34], i.e., in this case mice would subsequently be 6-8 weeks of age, when substantial neurodegeneration has already occurred. Related to this was our curious observation that CSTB protein expression was substantially higher in the brains of mice sacrificed at 9 versus 2 months (Fig. 2a, c, e). It appears that maximal expression required even more time than documented with other gene therapies, suggesting an additional reason to aim for early intervention in future studies [7].
In addition to the epilepsy, ataxia is a cardinal feature of EPM1 and is a highly disabling symptom. The cerebellum is the most affected region of the brain with neurodegeneration noted in both the Cstb−/− mice and human patients [35, 36]. Historically, prior to the availability of more modern anti-seizure drugs, the use of phenytoin to control seizures would result in accelerated cerebellar neurodegeneration and rapid loss of ambulation [37, 38]. Remarkably, in the present work, despite treatment at time points after late-into-neurodegeneration intervention, sufficient cerebellar cells were rescued and murine ataxia improved, suggesting that gene therapy will likely benefit patients even once the disease has already progressed beyond the earliest symptoms. Of course, newly diagnosed patients and genetically confirmed still asymptomatic siblings will likely benefit the most, as will cases detected very early through new-born screening in a hoped-for not too distant future.
Materials and methods
Study design
The experimental design for the study is summarized in Fig. 1b. Briefly, equal numbers of male and female mice were divided in three cohorts. For each cohort, CSTB-deficient (Cstb–/–) mice were injected with 5 μL of scAAV9/CSTB vector solution (7 ×1011 vg/mouse) or PBS via lumbar puncture. The first and second cohorts were injected at P21, while the third cohort received injections at P60. Cohort 1 consisted of 14 WT, 13 PBS injected and 13 AAV9 injected mice. Cohort 2 consisted of 12 WT, 10 PBS injected and 15 AAV9 injected mice. Cohort 3 consisted of 12 WT, 10 PBS injected and 15 AAV9 injected mice. Untreated (Cstb+/+) mice of the same background were used as wild type (WT) controls. At two months of age, cohort 1 was sacrificed to evaluate vector biodistribution, along with early biochemical and histological signs of treatment efficacy by measuring neuroinflammatory markers using qPCR and cerebellar granular cell death using TUNEL assay. All remaining mice were maintained to evaluate behavioral phenotype at seven to nine months of age. After rotarod testing, the mice were sacrificed to evaluate vector biodistribution, and treatment effects on late neuroinflammatory markers and neurodegeneration.
Plasmid construction and viral packaging
We designed and developed the CBh-hCSTB-bGHpA plasmid containing the transgene of a human CSTB codon-optimized construct. The CBh promoter was used to drive the expression of the human CSTB, and a 3’ bGH poly(A) signal was used to stabilize the transcript. The cassette was flanked by AAV2 inverted terminal repeats and cloned in a self-complementary fashion for enhanced post-transduction stability and expression (Fig. 1a). The final plasmid was packaged in AAV9 at the University of North Carolina (UNC) Vector Core facility, as described [39].
Mice
CSTB-deficient mice (Cstb−/−) were obtained from The Jackson Laboratory (129-Cstbtm1Rm/J) [6]. Wild type littermates, with intact Cstb gene (Cstb+/+), of the same age in each cohort were left untreated and used as controls. Both sexes were used in approximately equal proportions in all experiments. Samples sizes were chosen based on literature review of known effect size for rotarod analysis anticipating 50% rescue. All procedures were carried out according to NIH guidelines and the animal care committee regulations at the University of Texas Southwestern Medical Center. 7 ×1011 vector genomes or 5 μl PBS were injected intrathecally, as described [40]. Mice from alternative cages were used for each group for randomization. The person injective the mice was blinded to the selection of the animals and was involved in further analysis of the mouse. Behavioral studies were performed by a technician who was blinding to the genotype and the treatment group. Mice were sacrificed by cervical dislocation, and the brain was harvested and cut into two hemispheres. One hemisphere was fixed in formalin for paraffin embedding and histo- and immunohisto-chemistry. The other was snap-frozen in liquid nitrogen, ground into powder by mortar and pestle, and the powder divided into 30–40 mg aliquots in screwcap tubes for the genetic and biochemical experiments below.
Immunohistochemistry
Tissue sections (5 µm) were deparaffinized and rehydrated using xylenes and gradual concentration decreases of ethanol in water. Hydrated sections were then subjected to antigen retrieval by citrate buffer pH 6.0 (Sigma). Endogenous peroxidase activity was blocked for 10 min with BLOXALL solution (Vector labs). Sections were incubated with anti-cystatin B antibody (F-5) (Santa Cruz, sc-166561) diluted (1:100) in normal horse serum overnight at 4 °C, then successively incubated with Amplifier Antibody and ImmPRESS Polymer Reagent (Vector labs) and the ImmPACT DAB EqV working solution (Vector labs) until desired stain intensity.
Western blot
Ice cold RIPA lysis buffer (NaCl 150 mM, NP-40 1%, Sodium deoxycholate 0.5%, SDS 0.1%, Tris HCl 50 mM pH 8.0) containing protease inhibitor (ThermoScientific) was used to lyse and homogenize frozen ground brain tissue. Lysates were centrifuged at 14,000 × g for 5 min at 4 °C and supernatant was collected. Bradford assay reagent (ThermoScientific) was used to obtain Protein concentration. Equal amounts of whole protein from each lysate were subjected to SDS-PAGE electrophoresis TGX Stain-Free FastCast Acrylamide kit (BioRad) and then transferred to polyvinylidene difluoride (PVDF) membrane (Millipore) overnight at 4 °C. The membrane was probed with anti-Cystatin B (1:1000 dilution) (Santa Cruz Biotechnology, sc-101510). Protein densitometry was performed using Image Lab software (BioRad). The density of each protein band was normalized to the intensity of its corresponding whole protein image.
Quantitative real-time PCR
RNA was extracted using TriZol (Invitrogen) and purified using the PureLink RNA Mini Kit (Invitrogen). cDNA was synthesized by the iScript Reverse Transcription SuperMix kit (BioRad). The expression level of immune system related genes Cxcl1, Cxcl10, CxCl13, Gfap, and Iba1 was quantified by quantitative real-time PCR (qRT-PCR) using the QuantStudio 7 Pro System thermo-cycler (Thermo Scientific) and SYBR Green Master Mix (Bio-Rad). Data shows fold change relative to control samples using the ΔΔCq method with Rpl4 as an internal control gene. Primers are listed in Table 1.
Apoptosis assay
Mouse brain samples were paraffin embedded, processed, and sectioned according to standard procedures [41, 42]. Serial paraffin sections were concomitantly prepared and checked by dark field microscopy [43]. Resulting sections were stained by hematoxylin & eosin (H&E), and Terminal deoxynucleotidyltransferase-mediated UTP End Labeling (TUNEL). Confirmation of neuron cell death was performed via TUNEL; positive nuclei of apoptotic and necrotic cells were labeled with fluorescein according to methods of first report and literature supplied with the DeadEnd Fluorometric TUNEL System [44, 45]. Sections subjected to TUNEL were counterstained with propidium iodide. Stained slides were scanned using the Hamamatsu NanoZoomer 2.0 HT digital slide scanner (40 × objectives), and the number of positive nuclei in the cerebellum was quantified using HistoQuant (3DHistech).
Microscopy
Bright field images were taken by a Hamamatsu NanoZoomer 2.0-HT whole slide imaging system and fluorescence imaging was performed using Zeiss Axioscan.Z1 digital slide scanner in Whole Brain Microscopy Facility at UT Southwestern Medical Center at Dallas.
Behavioral study
Seven to nine month old mice were subjected to the rotarod test. Mice were placed on a stationary rotarod (Columbus Instruments, 3 cm diameter rod) and the latency to fall was recorded. The maximum time on the rod was 1 min. The stationary rod test was repeated 5 times with approximately 5–10 min between trials. The mice were then placed on a rod rotating slowly at 2 rpm. The latency to fall from the rod was recorded for each mouse. If a mouse held onto the rod and rotated completely around, it was treated as if it had fallen from the rod at that time. The maximum latency was again 1 minute. After two training trials, each mouse was tested three times with approximately 5–10 min between the experimental trials.
Statistical analysis
Student’s unpaired t-test was used to compare single means between two conditions. To assess the significance between three groups, one-way ANOVA followed by Tukey’s multiple comparisons test was performed. Data were analyzed and graphed using the GraphPad Prism software (v. 8.0.2; GraphPad Software). For all comparisons, statistical significance was set at p < 0.05. Asterisks denote level of significance based on p value: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Data availability
All raw data used to prepare this manuscript is available upon contacting corresponding author.
References
Crespel A, Ferlazzo E, Franceschetti S, Genton P, Gouider R, Kalviainen R, et al. Unverricht-Lundborg disease. Epileptic Disord. 2016;18:28–37.
Lalioti MD, Scott HS, Buresi C, Rossier C, Bottani A, Morris MA, et al. Dodecamer repeat expansion in cystatin B gene in progressive myoclonus epilepsy. Nature. 1997;386:847–51.
Joensuu T, Lehesjoki AE, Kopra O. Molecular background of EPM1-Unverricht-Lundborg disease. Epilepsia. 2008;49:557–63.
Mancini GM, Schot R, de Wit MC, de Coo RF, Oostenbrink R, Bindels-de Heus K, et al. CSTB null mutation associated with microcephaly, early developmental delay, and severe dyskinesia. Neurology. 2016;86:877–8.
O’Brien A, Marshall CR, Blaser S, Ray PN, Yoon G. Severe neurodegeneration, progressive cerebral volume loss and diffuse hypomyelination associated with a homozygous frameshift mutation in CSTB. Eur J Hum Genet. 2017;25:775–8.
Di Matteo F, Pipicelli F, Kyrousi C, Tovecci I, Penna E, Crispino M, et al. Cystatin B is essential for proliferation and interneuron migration in individuals with EPM1 epilepsy. EMBO Mol Med. 2020;12:e11419.
Pennacchio LA, Bouley DM, Higgins KM, Scott MP, Noebels JL, Myers RM. Progressive ataxia, myoclonic epilepsy and cerebellar apoptosis in cystatin B-deficient mice. Nat Genet. 1998;20:251–8.
Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. 2019;18:358–78.
Lykken EA, Shyng C, Edwards RJ, Rozenberg A, Gray SJ. Recent progress and considerations for AAV gene therapies targeting the central nervous system. J Neurodev Disord. 2018;10:16.
Shannon P, Pennacchio LA, Houseweart MK, Minassian BA, Myers RM. Neuropathological changes in a mouse model of progressive myoclonus epilepsy: cystatin B deficiency and Unverricht-Lundborg disease. J Neuropathol Exp Neurol. 2002;61:1085–91.
Franceschetti S, Sancini G, Buzzi A, Zucchini S, Paradiso B, Magnaghi G, et al. A pathogenetic hypothesis of Unverricht-Lundborg disease onset and progression. Neurobiol Dis. 2007;25:675–85.
Husi H, Fernandes M, Skipworth RJ, Miller J, Cronshaw AD, Fearon KCH, et al. Identification of diagnostic upper gastrointestinal cancer tissue type-specific urinary biomarkers. Biomed Rep. 2019;10:165–74.
Tegelberg S, Kopra O, Joensuu T, Cooper JD, Lehesjoki AE. Early microglial activation precedes neuronal loss in the brain of the Cstb−/− mouse model of progressive myoclonus epilepsy, EPM1. J Neuropathol Exp Neurol. 2012;71:40–53.
Sanz P, Serratosa JM. Neuroinflammation and progressive myoclonus epilepsies: from basic science to therapeutic opportunities. Expert Rev Mol Med. 2020;22:e4.
Okuneva O, Korber I, Li Z, Tian L, Joensuu T, Kopra O, et al. Abnormal microglial activation in the Cstb(−/−) mouse, a model for progressive myoclonus epilepsy, EPM1. Glia. 2015;63:400–11.
Okuneva O, Li Z, Korber I, Tegelberg S, Joensuu T, Tian L, et al. Brain inflammation is accompanied by peripheral inflammation in Cstb (−/−) mice, a model for progressive myoclonus epilepsy. J Neuroinflammation. 2016;13:298.
Manninen O, Koskenkorva P, Lehtimaki KK, Hypponen J, Kononen M, Laitinen T, et al. White matter degeneration with Unverricht-Lundborg progressive myoclonus epilepsy: a translational diffusion-tensor imaging study in patients and cystatin B-deficient mice. Radiology. 2013;269:232–9.
Manninen O, Laitinen T, Lehtimaki KK, Tegelberg S, Lehesjoki AE, Grohn O, et al. Progressive volume loss and white matter degeneration in cstb-deficient mice: a diffusion tensor and longitudinal volumetry MRI study. PLoS One. 2014;9:e90709.
Sipila JOT, Hypponen J, Kyto V, Kalviainen R. Unverricht-Lundborg disease (EPM1) in Finland: a nationwide population-based study. Neurology. 2020;95:e3117–e23.
Norio R, Koskiniemi M. Progressive myoclonus epilepsy: genetic and nosological aspects with special reference to 107 Finnish patients. Clin Genet. 1979;15:382–98.
Paulson H. Repeat expansion diseases. Handb Clin Neurol. 2018;147:105–23.
Nitschke F, Ahonen SJ, Nitschke S, Mitra S, Minassian BA. Lafora disease - from pathogenesis to treatment strategies. Nat Rev Neurol. 2018;14:606–17.
Alakurtti K, Weber E, Rinne R, Theil G, de Haan GJ, Lindhout D, et al. Loss of lysosomal association of cystatin B proteins representing progressive myoclonus epilepsy, EPM1, mutations. Eur J Hum Genet. 2005;13:208–15.
Zerovnik E. Human stefin B: from its structure, folding, and aggregation to its function in health and disease. Front Mol Neurosci. 2022;15:1009976.
Kopitar-Jerala N. The role of stefin B in neuro-inflammation. Front Cell Neurosci. 2015;9:458.
Penna E, Cerciello A, Chambery A, Russo R, Cernilogar FM, Pedone EM, et al. Cystatin B involvement in synapse physiology of rodent brains and human cerebral organoids. Front Mol Neurosci. 2019;12:195.
Daura E, Tegelberg S, Yoshihara M, Jackson C, Simonetti F, Aksentjeff K, et al. Cystatin B-deficiency triggers ectopic histone H3 tail cleavage during neurogenesis. Neurobiol Dis. 2021;156:105418.
Xu TT, Zeng XW, Wang XH, Yang LX, Luo G, Yu T. Cystatin-B negatively regulates the malignant characteristics of oral squamous cell carcinoma possibly via the epithelium proliferation/differentiation program. Front Oncol. 2021;11:707066.
Lehtinen MK, Tegelberg S, Schipper H, Su H, Zukor H, Manninen O, et al. Cystatin B deficiency sensitizes neurons to oxidative stress in progressive myoclonus epilepsy, EPM1. J Neurosci. 2009;29:5910–5.
Ceru S, Konjar S, Maher K, Repnik U, Krizaj I, Bencina M, et al. Stefin B interacts with histones and cathepsin L in the nucleus. J Biol Chem. 2010;285:10078–86.
Chakrabarty P, Rosario A, Cruz P, Siemienski Z, Ceballos-Diaz C, Crosby K, et al. Capsid serotype and timing of injection determines AAV transduction in the neonatal mice brain. PLoS One. 2013;8:e67680.
Hollidge BS, Carroll HB, Qian R, Fuller ML, Giles AR, Mercer AC, et al. Kinetics and durability of transgene expression after intrastriatal injection of AAV9 vectors. Front Neurol. 2022;13:1051559.
Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16:1073–80.
Reimsnider S, Manfredsson FP, Muzyczka N, Mandel RJ. Time course of transgene expression after intrastriatal pseudotyped rAAV2/1, rAAV2/2, rAAV2/5, and rAAV2/8 transduction in the rat. Mol Ther. 2007;15:1504–11.
Chew NK, Mir P, Edwards MJ, Cordivari C, Martino D, Schneider SA, et al. The natural history of Unverricht-Lundborg disease: a report of eight genetically proven cases. Mov Disord. 2008;23:107–13.
Magaudda A, Ferlazzo E, Nguyen VH, Genton P. Unverricht-Lundborg disease, a condition with self-limited progression: long-term follow-up of 20 patients. Epilepsia. 2006;47:860–6.
Eldridge R, Iivanainen M, Stern R, Koerber T, Wilder BJ. “Baltic” myoclonus epilepsy: hereditary disorder of childhood made worse by phenytoin. Lancet. 1983;2:838–42.
Iivanainen M, Eldridge R. Effect of phenytoin on the mental and physical function of patients with Baltic myoclonus epilepsy. Ital J Neurol Sci. 1987;8:313–7.
Clement N, Grieger JC. Manufacturing of recombinant adeno-associated viral vectors for clinical trials. Mol Ther Methods Clin Dev. 2016;3:16002.
Gray SJ, Choi VW, Asokan A, Haberman RA, McCown TJ, Samulski RJ. Production of recombinant adeno-associated viral vectors and use in in vitro and in vivo administration. Curr Protoc Neurosci. 2011;Chapter 4:Unit 4 17.
Sheehan DC, Hrapchak BB. Theory and practice of histotechnology, 2nd ed. Mosby; 1980.
Woods AEAE, Roy C. Laboratory histopathology, a complete reference. Churchill-Livingston Press; 1996.
Shelton JM, Grauer G, Richardson JA. Combination low magnification dark-field illuminator and bright-field microscopy substage condenser with descriptions and modifications to manufacture this device from commercially available fiber-optic ring lights, UTSD: 1494. 2003. http://www.utsouthwestern.edu/labs/molecular-pathology/about/intellectualproperty/utsd1494.html.
Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501.
Promega Technical Bulletin 235 (TB235). Instruction Booklet for Use of Product G3250. Madison WI, USA: Promega Corporation; 2008.
Acknowledgements
This work was funded by Ultragenyx and Hope for ULD. We thank the UT Southwestern Histopathology (Dr. John Shelton), Whole Brain Microscopy (Dr. Denise Ramirez), Rodent Behavior core (Dr. Shari Birnbaum) and Neuro-Models facility (Dr. Erik Plautz) for assistance with tissue processing, imaging, the rotarod test and video-electrocorticography, respectively. BAM holds the University of Texas Southwestern Jimmy Elizabeth Westcott Chair in Pediatric Neurology.
Author information
Authors and Affiliations
Contributions
BAM and EG designed the project. BAM wrote the manuscript. EG, SK, and MV conducted the experiments, analyzed the data, and helped write the manuscript. DVA, UM, and XC helped in parts of experiments. SFM, AL, and SJG provided feedback and reviewed the manuscript. BAM supervised the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gumusgoz, E., Kasiri, S., Verma, M. et al. CSTB gene replacement improves neuroinflammation, neurodegeneration and ataxia in murine type 1 progressive myoclonus epilepsy. Gene Ther (2023). https://doi.org/10.1038/s41434-023-00433-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41434-023-00433-x
